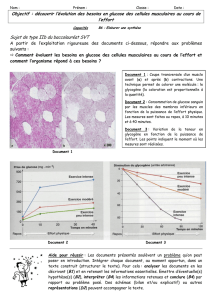
Glut1 deficiëntie werd voor het eerst beschreven in 1991 door De Vivo

Maladie de De Vivo ou syndrome de déficit en GLUT-1
Déficit sous-jacent :
Le déficit en Glut-1 a été décrit pour la première fois en 1991 par De Vivo, à New York. Le problème
provoqué par un déficit en Glut-1 est que le glucose (sucre sanguin) n'est pas correctement transporté
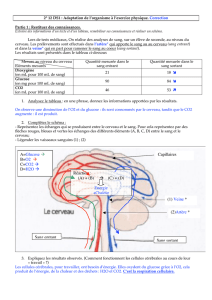
vers le cerveau. Le GLUT-1 est la protéine qui transporte le glucose au-delà de la barrière hémato-
encéphalique (BHE). La BHE est une barrière présente dans le cerveau entre la circulation sanguine
et le système nerveux central. Le glucose est le carburant du cerveau. C'est pourquoi un déficit dans
son transport, comme en GLUT-1, donne lieu à un déficit en énergie pour le cerveau.
La protéine GLUT-1 est codée sur le gène SLC2A1 (transporteur du glucose de type 1). La protéine
fait partie des membranes cellulaires, GLUT-1 étant impliquée dans le transport du glucose au travers
de la barrière hémato-encéphalique. Les mutations du gène SLC2A1 réduisent l'activité de la GLUT-1,
ce qui réduit donc l'apport en glucose au cerveau.
Syndrome :
Généralement, des crises d'épilepsie se manifestent entre 1 et 4 mois. L'épilepsie est souvent plus
grave si le patient n'a rien mangé depuis longtemps et la réaction à l'ingestion de glucides est bonne.
Le patient souffrant de crises ne réagit pas bien aux médicaments utilisés dans le traitement de
l'épilepsie. De plus, les enfants peuvent souffrir de brèves crises de mouvements des yeux verticaux,
horizontaux ou circulaires. De plus, on assiste à un retard de développement et à des problèmes
d'apprentissage.
Les enfants ont également souvent une petite tête. De plus, ils peuvent souffrir d'un trouble de la
coordination des mouvements volontaires (ataxie) et d'une baisse de tension du muscle (hypotonie). Il
peut également être question de spasticité.
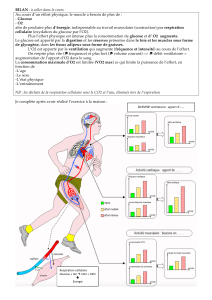
Le glucose sanguin est normal, le taux de glucose dans le cerveau étant par contre fortement réduit.
D'autres symptômes peuvent se manifester, comme confusion, fatigue, paralysie d'un côté du corps,
problèmes de sommeil et maux de tête.
On distingue 3 phénotypes (symptômes extrêmes) :
1. Syndrome de déficit en Glut1 classique
Parmi les symptômes, citons retard de développement et épilepsie. Les crises d'épilepsie
commencent avant l'âge de 4 mois : toutes les formes d'épilepsie se manifestent. On assiste
également à des périodes de mouvements rapides des yeux. Le nombre de crises d'épilepsie varie
d'un patient à l'autre. Certains patients souffrent de crises d'épilepsie quotidiennes, d'autres une fois
par jour, par semaine ou par mois. Cependant, les patients ne réagissent pas bien aux médicaments
utilisés contre l'épilepsie et se sentent rapidement mieux après avoir commencé une diète cétogène.
Les troubles neurologiques (comme confusion, mouvements anormaux, paralysie, problèmes de
sommeil et maux de tête récurrents) varient fortement et dépendent notamment de périodes de jeûne
et de fatigue. Les patients éprouvent des difficultés à parler et avec le développement de la langue. Le
développement cognitif varie d'un patient à l'autre et va de problèmes d'apprentissage à un grave
handicap mental. Chez la plupart des patients, la maladie n'est pas progressive, bien que notamment
les troubles de coordination des mouvements (ataxie ou spasticité) puissent s'aggraver au fil du
temps, surtout si le patient n'est pas traité à l'aide d'une diète cétogène (voir traitement.)
2. Syndrome de déficit en Glut-1 non-classique
Handicap mental et démarche chancelante (spasticité), mais sans crises d'épilepsie.
Problématique motrice et neurologique complexe : mouvements soudains, coordonnés et
involontaires,de flexion et d'extension lentes des doigts et orteils (choréoathétose), myoclonies,
trémulation, tension musculaire changeante (dystonie ; parkinsonisme), ataxie, dyspraxie et soudaine
perte de force musculaire.
Les adultes sont touchés par des problèmes moteurs mineurs.

Attention en cas d'effort musculaire excessif, vu qu'il peut donner lieu à une aggravation des
symptômes neurologiques, mais également à une destruction des globules rouges (anémie
hémolytique).
Diagnostic :
Le diagnostic est posé à l'aide d'un examen clinique et d'un examen du liquide cérébro-spinal. Une
ponction lombaire est nécessaire pour l'examen du liquide cérébro-spinal. Dans le liquide cérébro-
spinal, les concentrations de glucose et de lactate sont en effet basses, alors que les valeurs du taux
de glucose dans le sang sont normales : le ratio du liquide cérébro-spinal/glucose dans le sang = <
0,5 (normalement > 0,6). Pour le diagnostic définitif du déficit en glut-1, un examen du GLUT-1 dans
les globules rouges (examen de la captation du glucose) et/ou un examen d'hérédité du gène SLC2A1
est nécessaire. Une IRM et un CT scan du cerveau sont généralement normaux.
Un examen prénatal est également possible si l'anomalie génique du membre de la famille est
connue.
Traitement :
Le déficit en Glut-1 ne se guérit pas. Pour contrôler les crises d'épilepsie, une diète cétogène est
prescrite. Il s'agit d'un régime très riche en graisses où la plupart des glucides sont remplacés par de
la graisse. La diète induit également la consommation de très peu de protéines. Une diète cétogène
doit être prescrite par un médecin expérimenté dans ce domaine et faire l'objet d'un encadrement par
un diététicien expérimenté. Grâce à une diète très riche en graisses, l'organisme produit des corps
cétoniques, qui normalement ne sont produits qu'en cas de jeûne de longue durée. Les corps
cétoniques servent de carburant alternatif au cerveau. La diète a un effet positif sur le développement
cognitif et les troubles du comportement.
L'acide alpha-lipoïque (acide thioctique) est un antioxydant qui contribue également au transport du
glucose, notamment dans des études in vitro (dans des cellules). Jusqu'à présent, les résultats sont
modérés, probablement car le dosage administré par voie orale est trop faible. Il est préférable
d'attendre la publication d'autres résultats.
Les médicaments à éviter sont les barbituriques (par ex. le phénobarbital et le diazépam sont prescrits
pour les enfants souffrant d'épilepsie, mais aggravent l'état de l'enfant) et les méthylxanthines
(empêchent le transport de glucose). Les méthylxanthines sont également présentes dans le café et
d'autres produits à base de caféine.
Voici les antiépileptiques qui sont autorisés : carbamazépine, phénytoïne, zonisamide et (valproate).
Héritabilité :
Autosomique récessif ou autosomique dominant.
1
/
2
100%