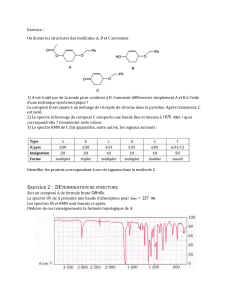
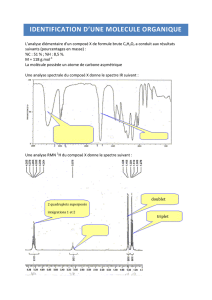
Composés aromatiques : Spectroscopie IR et RMN

Complément - Chapitre 8
Composés aromatiques
Caractéristiques spectrales des composés aromatiques
- Spectroscopie infrarouge (IR)
Les bandes d’absorption des composés aromatiques en sp ectroscopie infrarouge sont
similaires à celles des alcènes, puisque ce sont des composés insaturés formés de
liaisons C=C. Le tableau 8.a illustre celles en IR les plus fréquemment observées. Ce
sont des bandes d’absorption d’intensité variable dues à l’élongation des liaisons C–H
(3100 cm-1-3000 cm-1) et C=C (1600 cm-1-1450 cm-1).
Tableau 8.a Bandes IR caractéristiques des composés aromatiques
Type de liaison Nombre d’onde
(cm-1) Mode de vibration Intensité
C–H (aromatique) 3 100-3 000 Élongation Variable (moyenne
à faible)
C=C (aromatique) 1 600-1 450 Élongation Variable (forte à
moyenne)
L’analyse de l’empreinte digitale (entre environ 850 cm-1 et 600 cm-1) s’avère fort
intéressante ; les bandes d’absorption sont assez intenses et elles représentent les
vibrations des déformations angulaires hors du plan des liaisons C–H. En fait, elle
permet de déduire si le composé aromatique étudié est substitué (monosubstitué, di-,
tri-, tétra-, etc.) et de déterminer la position des substituants, par exemple les positions
ortho, méta ou para dans le cas d’un composé aromatique disubstitué. Le tableau 8.b.
présente les bandes IR des déformations angulaires hors du plan des liaisons C–H
correspondant à quelques possibilités de composés aromatiques substitués.
Tableau 8.b Bandes d’absorption IR des déformations angulaires hors du plan
des liaisons C–H caractéristiques de quelques composés aromatiques substitués
Substitution du cycle aromatique Nombre d’onde
(cm-1)
Composé aromatique monosubstitué 770-730 et ∼700
Composé aromatique disubstitué
(en position ortho) 770-730
Composé aromatique disubstitué
(en position méta) 800-750 et ∼700
Composé aromatique disubstitué
(en position para) 850-800
Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 1

On constate au tableau 8.b que les bandes IR des liaisons C–H au niveau de
l’empreinte digitale sont relativement près les unes des autres. Les spectres
infrarouges sont souvent difficiles à interpréter. De ce fait, il faut effectuer plusieurs
caractérisations (RMN 1H, spectre de masse…) d’un composé étudié, en plus du
spectre infrarouge, pour pouvoir apporter des conclusions avec certitude quant à sa
nature.
Dans les spectres IR du benzène et du o-xylène illustrés à la figure 8.a, les bandes IR
des composés aromatiques caractéristiques sont observées.
Figure 8.a Spectres IR
a) du benzène
Spectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100
par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.
Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 2

b) du o-xylène
Spectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100
par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.
c) du m-xylène
Spectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100
par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.
Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 3

d) et du p-xylène
Spectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100
par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.
- Résonance magnétique nucléaire (spectre RMN)
Le cône d’anisotropie crée, dans le cas du cycle benzénique, un très grand déblindage
au niveau des hydrogènes directement liés au cycle. Ceux-ci résonnent alors à un
champ très faible et ont donc des déplacements chimiques élevés, soit entre 6,6 ppm
et 8,0 ppm. Par exemple, les hydrogènes du benzène, sans substituant, possèdent un
déplacement chimique de 7,27 ppm. Leur signal est un singulet, les six hydrogènes
étant équivalents (voir la figure 8.b).
Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 4

Figure 8.b Spectre RMN 1H du benzène (solvant : CDCl3)
Spectre réalisé sur Bruker Avance 400 PJAB par François Raymond, étudiant
au doctorat au laboratoire du professeur James D. Wuest à l'Université de Montréal.
Les spectres RMN 1H des composés aromatiques sont souvent assez complexes à
analyser. Une de leur particularité est qu’ils révèlent des couplages entre des
hydrogènes plutôt éloignés. On les distingue les uns des autres par leurs constantes de
couplage (J) d’intensité décroissante. Ainsi, les hydrogènes en position ortho ont des
constantes de couplage variant entre 6 Hz et 8 Hz et ceux en position méta en ont
d’environ 1 Hz à 3Hz. Les constantes de couplage des hydrogènes en position para
sont très faibles, voire inexistantes, allant de 0 Hz à 1 Hz.
De plus, les substituants activants (qui enrichissent le cycle aromatique en électrons)
ont un effet blindant tandis que les substituants désactivants (qui appauvrissent le
cycle aromatique en électrons) ont un effet déblindant sur les déplacements chimiques
des hydrogènes benzéniques. Les hydrogènes situés en position ortho du substituant
subissent davantage l’effet de ce dernier. Le tableau 8.c présente une approximation
de l’effet des différents substituants sur les hydrogènes benzéniques en fonction de
leur position. Les hydrogènes d’un cycle benzénique ne seront donc pas tous
équivalents selon la nature et le nombre de substituants sur le cycle, ce qui nécessite
alors une analyse plus approfondie, les spectres étant plus compliqués.
Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 5
 6
7
8
9
6
7
8
9
1
/
9
100%