Information sous embargo jusqu`au jeudi 16 octobre – 20h

1
Information sous embargo jusqu’au jeudi 16 octobre – 20h
Paris, le 16 octobre 2003
COMMUNIQUE DE PRESSE
Thérapie Génique du déficit immunitaire DICS-X :
Résultats des premières investigations
sur la complication survenue chez deux enfants traités
Alain Fischer, Marina Cavazzana-Calvo et Salima Hacein-Bey Abina (Unité Inserm 429,
Développement normal et pathologique du système immunitaire, Hôpital Necker Enfants
Malades, Paris) publient, dans la revue Science à paraître le 17 octobre 2003, les résultats
des investigations, fruits d’une collaboration internationale, concernant la complication
survenue chez deux des dix enfants traités par thérapie génique pour le déficit immunitaire
combiné sévère lié à l’X (DICS-X). Les chercheurs indiquent que la prolifération incontrôlée
de lymphocytes observée est due à la surexpression d’un oncogène∗∗
∗∗ dans lequel s’est
inséré le vecteur rétroviral transportant le gène-médicament. Certains facteurs
permettraient d’expliquer la fréquence inattendue de cette complication : spécificité des
cellules traitées et de la maladie, âge des deux enfants au moment de la thérapie génique…
Ces travaux d’investigation ont été effectués grâce au soutien de l’AFM, à travers les dons
du Téléthon, à hauteur de 145 000 euros. Les chercheurs poursuivent leurs travaux afin de
définir des critères de « sécurisation» pour l’application ultérieure de cette stratégie
thérapeutique. En effet, plus de quatre ans après le traitement du premier patient, la
thérapie génique s’avère toujours efficace puisque le système immunitaire des enfants
traités est fonctionnel.
Dans le cadre d’un essai de thérapie génique débuté en 1999 et dont le promoteur est l’AP-HP,
Alain Fischer, Marina Cavazzana-Calvo, Salima Hacein-Bey Abina et leur équipe sont parvenus à
corriger un déficit immunitaire héréditaire chez neuf « enfants-bulle » (sur les dix enfants inclus
dans l’essai). Au cours de l’année 2002, une complication est survenue chez deux d’entre eux
conduisant à une suspension transitoire de cet essai. Des recherches ont été entreprises pour
comprendre les raisons de cette complication et re-évaluer le rapport bénéfice-risque de cette
thérapie pour le déficit immunitaire DICS-X.
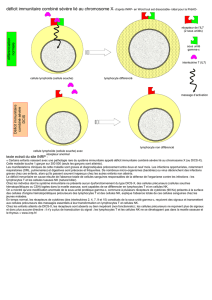
Au terme de plusieurs mois d’investigation, les chercheurs démontrent que la prolifération clonale
de lymphocytes T observée chez les deux enfants est due à l’insertion au sein d’un proto-
oncogène, LMO2, situé sur le chromosome 11, du vecteur rétroviral transportant le gène-
médicament dans les cellules souches hématopoïétiques. LMO2 code une protéine impliquée
dans la régulation de l’hématopoïèse (production des cellules sanguines) dont la surexpression a
∗ gène responsable du développement tumoral.

2
été l’élément déclencheur du processus menant à la prolifération incontrôlée de lymphocytes. Pour
enrayer ce phénomène pathogène, les deux enfants concernés ont reçu un traitement par
chimiothérapie et une greffe de moelle osseuse pour l’un d’entre eux. La prolifération a été
maîtrisée et les deux enfants se portent bien, avec, chez l’un la disparition totale de cellules
anormales et, chez l’autre, la présence résiduelle de quelques-unes de ces cellules.
Ce risque potentiel de « mutagenèse insertionnelle », qui n’avait pas été observé dans les études
pré-cliniques, semble s’être avéré plus fréquent que prévu. Trois hypothèses sont envisagées par
les chercheurs :
¾ Une plus grande fréquence d’insertion du vecteur au sein de zones « actives » du génome,
notamment le gène LMO2.
¾ Un risque inhérent à la maladie elle-même : chez les malades atteints de DICS-X, le nombre
de cellules précurseurs à capacité proliférative potentielle pourrait être plus élevé que la
normale. Il y aurait donc un plus grand nombre de cellules cibles potentielles d’un tel
évènement.
¾ L’âge des patients : les deux enfants concernés étaient les plus jeunes lors du traitement :ils
étaient respectivement âgés de 1 et 3 mois, une période où le nombre de cellules précurseurs
cibles de la thérapie génique pourrait être plus élevé .
Reste aujourd’hui aux chercheurs à évaluer ces hypothèses, à travers notamment la mise au point
de modèles animaux. Par ailleurs, ils poursuivent, chez tous les malades traités, le travail de
caractérisation de l’ensemble des sites d’insertion du vecteur rétroviral dans les lymphocytes.
Compte-tenu de l’efficacité démontrée de la thérapie génique pour ce déficit immunitaire
héréditaire, ces recherches sont cruciales : elles permettront d’apporter les modifications
nécessaires à la reprise de l’essai en limitant les risques pour les malades au regard des
bénéfices indubitables apportés par cette stratégie thérapeutique.
Le déficit immunitaire combiné sévère lié au chromosome X est une maladie génétique héréditaire
rare caractérisée par une absence totale de cellules responsables de la défense de l’organisme
contre les infections, les lymphocytes T et les cellules NK (natural killer).
Publication
« LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-
X1 » S. Hacein-Bey-Abina, C. Von Kalle, M. Schmidt, M.P. McCormack, N. Wulffraat, P. Leboulch,
A. Lim, C.S. Osborne, P. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P. Fraser, J.L. Cohen, G.
de Saint-Basile, I. Alexander, U. Wintergerst, T. Frebourg, A. Aurias, D. Stoppa-Lyonnet, S.
Romana, I. Radford-Weiss, F. Gross, F. Valensi, E. Delabesse, E. MacIntyre, F. Sigaux, J. Soulier,
L.E. Leiva, M. Wissler, C. Prinz, T. H. Rabbitts, F. Le Deist, A. Fischer, M. Cavazzana-Calvo.
Science, 17 octobre 2003.
Contacts Presse
AFM
Emmanuelle Guiraud
Estelle Assaf
Tél : 01 69 47 28 28
AP-HP
Thierry Girouard
Tél : 01 40 27 37 22
paris.fr
Inserm
Séverine Ciancia
Tél : 01 44 23 60 86
1
/
2
100%