Maladie de Creutzfeldt-Jakob iatrogène et - iPubli

JO : 7
M
.
n°
<<

vol. 94
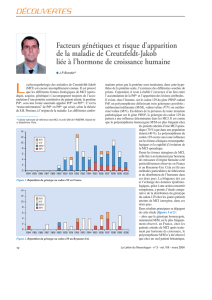
••• BRÈVES •••
K+
F F
1
/
2
100%