Lire l`article complet

DOSSIER
La Lettre du Cardiologue - n° 325 - février 2000
23
organisme dispose d’un puissant système de métabo-
lisation des médicaments, situé principalement dans le
foie, qui les “détoxifie” en les rendant hydrosolubles
et/ou en les conjuguant pour permettre leur élimination dans
l’urine ou les fèces. Les réactions de métabolisation hépatique
comprennent des réactions dites de “phase 1” (oxydation, réduc-
tion, hydrolyse principalement) et des réactions de phase 2 (sulfo-
ou glucuronoconjugaison). Les réactions d’oxydation sont sous
la dépendance d’un système enzymatique complexe : celui des
cytochromes P450 (CYP), enzymes membranaires qui catalysent
les réactions de mono-oxygénation. Ils sont localisés préféren-
tiellement dans le foie, mais aussi dans l’épithélium de l’intestin
grêle. Plusieurs isoformes de CYP ont été identifiés et un sys-
tème de nomenclature permet de les différencier. Ils sont grou-
pés en familles et sous-familles. Seules les familles CYP1, CYP2
et CYP3 et leurs sous-familles : CYP2D6, CYP3A4, CYP1A2,
CYP2C9, CYP2C19 présentent un intérêt clinique dans le méta-
bolisme des médicaments. Certains CYP, tel le CYP2C19, sont
des enzymes soumises au polymorphisme génétique : dans la
population, on retrouve ainsi des phénotypes enzymatiques
“intensifs”, “intermédiaires” ou “pauvres” qui induisent des varia-
tions du métabolisme médicamenteux considéré.
Une parfaite connaissance des réactions de biotransformation des
médicaments est utile pour prévoir de possibles interactions médi-
camenteuses : des études préalables peuvent être réalisées soit in
vitro (hépatocytes isolés, microsomes humains), soit in vivo. Pour-
tant, un certain nombre d’interactions médicamenteuses ne sont
découvertes que lors de notifications spontanées des médecins.
La maîtrise expérimentale du système des CYP est difficile. En
effet, un même CYP peut avoir deux médicaments pour substrats,
avec une affinité différente (tableau I) ; un CYP peut aussi induire
Les interactions médicamenteuses :
rôle des cytochromes P450
●B. Baldin*, A. Spreux*, M. Drici**
■
Par leur effet inhibiteur enzymatique du CYP450, des
médicaments très divers peuvent, lorsqu’ils sont asso-
ciés à d’autres, conduire à des effets toxiques délétères.
■
Les conséquences cliniques de cette inhibition dépen-
dent de plusieurs facteurs tels que la dose, la chronolo-
gie et la susceptibilité individuelle.
■
Les médicaments inducteurs du CYP450 stimulent le
métabolisme d’autres substrats et peuvent faire conclure,
à tort, à l’inefficacité des médicaments associés.
Mots-clés : Cytochrome P450 - Médicament - Inhibition
enzymatique - Induction enzymatique.
Points forts
*Centre régional de pharmacovigilance, hôpital Pasteur, Nice.
** Laboratoire de pharmaco-toxicologie, hôpital Pasteur, Nice.
L
‘
Tableau I. Principaux produits métabolisés par le CYP450 3A4.
Alfentanil
Alprazolam
Amiodarone
Amitriptyline
Amlodipine
Atorvastatine
Carbamazépine
Cérivastatine
Cisapride
Citalopram
Clarithromycine
Clopidogrel
Clozapine
Codéine
Colchicine
Cyclophosphamide
Ciclosporine A
Dapsone
Delavirdine
Dextrométhorphane
Diazépam
Diltiazem
Docétaxel
Ebastine
17-bêta-estradiol
Érythromycine
Éthinylestradiol
Félodipine
Finastéride
Flutamide
Gestodène
Granisétron
Halopéridol
Ifosfamide
Imipramine
Indinavir
Ivermectine
Lansoprazole
Lidocaïne
Loratadine
Meloxicam
Midazolam
Névirapine
Nicardipine
Nifédipine
Nimodipine
Nitrendipine
Oméprazole
Paclitaxel
Progestérone
Propafénone
Quinidine
Ritonavir
Saquinavir
Simvastatine
Sulfaméthoxazole
Sufentanil
Tacrolimus
Tamoxifène
Théophylline
Triazolam
Vérapamil
Warfarine

ou inhiber l’activité d’une autre sous-famille de cytochromes sans
en être le substrat (1). Enfin, un même médicament peut être méta-
bolisé par plusieurs CYP, avec des voies préférentielles qui sont
variables en fonction du polymorphisme génétique.
INHIBITION ENZYMATIQUE DES CYP :
SOURCE D’INTERACTIONS MÉDICAMENTEUSES
L’inhibition enzymatique est le mécanisme le plus souvent impli-
qué dans les interactions médicamenteuses (tableau II). Lorsque
deux substances sont métabolisées par le même cytochrome, elles
entrent en compétition. Le médicament ayant la plus forte affi-
nité pour le CYP occupe les sites de liaison compétitivement et
réduit la capacité du foie à métaboliser l’autre substance. Celle-
ci va s’accumuler dans l’organisme. Ce type d’interaction, qui
provoque une élévation des concentrations du médicament inhibé,
induit une réponse pharmacologique plus importante, entraînant
ainsi une majoration du risque d’effets indésirables, qui peut par-
fois mettre en jeu le pronostic vital.
✔Un des exemples les plus marquants en cardiologie est le cas
du mibéfradil (Posicor®), qui a été récemment retiré du marché.
Le mibéfradil est un puissant inhibiteur du CYP3A4, qui est une
des voies de biotransformation de nombreux médicaments, et
notamment de la majorité des “statines”. Ainsi, lors de la prise
concomitante de simvastatine, il existe un risque accru de rhab-
domyolyses sévères. De même, son association à des médica-
ments qui prolongent la durée de l’intervalle QT est susceptible
de faciliter l’apparition de torsades de pointe.
✔Un effet particulier du jus de pamplemousse est à signaler. Cer-
tains flavonoïdes du pamplemousse inhibent le CYP3A4 intesti-
nal et peuvent multiplier par dix la concentration plasmatique de
certains médicaments métabolisés par le même CYP (tableau I).
CARACTÉRISTIQUES DE L’INHIBITION ENZYMATIQUE
Chronologie
L’inhibition enzymatique débute dès que des concentrations suf-
fisantes de l’inhibiteur sont atteintes dans le foie (habituellement
en quelques heures) ; elle est maximale dans les 24 heures. Dès
l’arrêt du médicament inhibiteur, l’inhibition devient réversible
dans des délais qui varient selon la demi-vie du médicament inhi-
biteur (2, 3).
Ainsi, l’amiodarone (Cordarone®) est un agent inhibiteur du méta-
bolisme enzymatique hépatique des anticoagulants oraux. L’effet
inhibiteur de l’amiodarone se manifeste en général après une
semaine de traitement, atteint son niveau maximal après un mois
et disparaît quelques mois après son arrêt, compte tenu de sa longue
demi-vie (4). En conséquence, des cas d’hypoprothrombinémie
(variations de 50 à 100 %) et d’épisodes hémorragiques ont été
décrits au décours de sa coprescription avec la warfarine (4).
Dose
L’inhibition enzymatique est généralement dose-dépendante. La
cimétidine est un puissant inhibiteur enzymatique du CYP3A4
uniquement à une posologie supérieure ou égale à 800 mg par
jour ; aucune interaction n’a été mise en évidence à une posolo-
gie plus faible (5, 6).
Le fluconazole (Triflucan®) ne présente une action inhibitrice
enzymatique du CYP3A4 qu’à une posologie de 100 mg/j, voire
même 200 mg/j (7).
Susceptibilité individuelle
Certains patients peuvent présenter une déficience génétique de
certains cytochromes et seront donc plus sensibles que d’autres
aux inhibiteurs enzymatiques. Le CYP2D6 transforme la codéine
en morphine ; il en résulte que l’administration de quinidine, puis-
sant inhibiteur de ce CYP2D6, va entraîner une inefficacité de la
La Lettre du Cardiologue - n° 325 - février 2000
24
DOSSIER
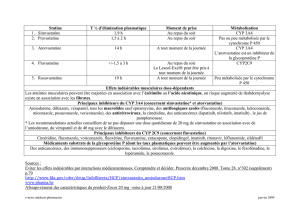
Tableau II. Inducteurs et inhibiteurs enzymatiques. (D’après la liste de D.A. Flockart) (http://www.dml.georgetour.edu/depts/pharmacology/davetab.html).
1A2 2C19 2C9 2D6 2E1 3A4
INHIBITEURS
Cimétidine Cimétidine Amiodarone Amiodarone Disulfirame Amiodarone
Ciprofloxacine Fluoxétine Fluconazole Fluoxétine Cimétidine
Fluvoxamine Fluvoxamine Fluoxétine Halopéridol Clarithromycine
Ofloxacine Kétoconazole Isoniazide Indinavir Érythromycine
Ticlopidine Lansoprazole Paroxétine Paroxétine Jus de pamplemousse
Oméprazole Ticlopidine Quinidine Itraconazole
Paroxétine Ritonavir Kétoconazole
Ticlopidine Sertraline
INDUCTEURS
Tabac Carbamazépine Phénobarbital Alcool Carbamazépine
Oméprazole Rifampicine Isoniazide Glucocorticoïdes
Griséofulvine
Phénytoïne
Rifampicine
Ritonavir
Absent chez 15 à 30 % Absent chez 1 % Absent chez 7 % Hyperactif chez 1 %
des patients des patients des patients des patients
de type asiatique de type caucasien de type caucasien de type caucasien

codéine chez les patients qui présentent un déficit en CYP2D6 (8 %
de la population de type caucasien, 2 % de la population de type
asiatique) (8).
Puissance inhibitrice
Elle varie selon les médicaments au sein d’une même classe. Dans
la famille des bloqueurs des canaux calciques, le diltiazem, le
vérapamil et la nicardipine apparaissent comme des inhibiteurs
enzymatiques de puissance modérée. Toutefois, le diltiazem et le
vérapamil multiplient les concentrations de ciclosporine par 2 à
4, risquant ainsi de majorer la toxicité rénale de la ciclosporine
(9, 10). Cet effet est d’autant plus préoccupant que ces médica-
ments peuvent être prescrits dans l’HTA résultant de l’adminis-
tration de ciclosporine. D’autres bloqueurs des canaux calciques
tels que l’isradipine, la nitrendipine et l’amlodipine ne semblent
pas modifier la ciclosporinémie.
De manière similaire, un risque de sédation prolongée peut être
provoqué par la coprescription de bloqueurs des canaux calciques
avec du midazolam ou de l’alfentanil (11).
Le vérapamil inhibe aussi le métabolisme de la quinidine, expo-
sant le patient à une majoration des effets indésirables (hypoten-
sion artérielle, bloc auriculoventriculaire...) (12).
Quelques interactions en cardiologie
Statines et antifongiques imidazolés. L’itraconazole et le kéto-
conazole sont de puissants inhibiteurs du CYP450. Leur associa-
tion à la simvastatine, la cérivastatine ou l’atorvastatine est contre-
indiquée en raison d’une élévation de leurs concentrations
plasmatiques qui induit un risque
accru de rhabdomyolyse (13).
Flécaïnide et amiodarone. L’inhi-
bition par l’amiodarone (400 mg/j)
du métabolisme de l’acétate de flé-
caïnide (100 mg) peut être respon-
sable d’une élévation majeure des
concentrations de cette dernière,
dont l’index thérapeutique est très
étroit. Cette interaction, bien que
très rare, est de mauvais pronostic,
avec apparition de blocs auriculo-
ventriculaires, de collapsus et
risque de mortalité élevé. Lorsque
l’association amiodarone-flécaï-
nide s’avère nécessaire, une réduction de 30 à 50 % de la posolo-
gie d’acétate de flécaïnide, associée à une surveillance biologique
(dosage plasmatique), est préconisée (14).
Bêtabloquants et lidocaïne. Le propranolol (Avlocardyl®...), le
métoprolol (Lopressor®,Seloken®...) et le nadolol (Corgard®)
administrés au long cours sont susceptibles d’inhiber le métabo-
lisme de la lidocaïne administrée en i.v. La toxicité résultante liée
à la lidocaïne (15) peut se manifester par des bradycardies sévères,
des arrêts cardiaques, ou encore des tableaux d’agitation et
d’agressivité. Si une telle association est nécessaire, il faut adap-
ter la posologie de la lidocaïne en fonction d’un contrôle des
concentrations plasmatiques et d’une surveillance clinique et élec-
trocardiographique des plus soigneuses.
Cisapride et macrolides, cisapride et imidazolés antifon-
giques. Le cisapride (Prepulsid®) est un médicament pouvant être
responsable de l’allongement de l’intervalle QT avec risque de
torsades de pointe. Ce risque est majoré en cas d’inhibition de
son métabolisme par les macrolides (tous, sauf la spiramycine)
ou par les imidazolés antifongiques (itraconazole, fluconazole,
miconazole) : ces associations sont contre-indiquées (16).
INDUCTION ENZYMATIQUE
Certains médicaments augmentent l’activité catalytique du
CYP : l’induction enzymatique affecte principalement la
phase I du métabolisme (oxydation, réduction, hydrolyse), et à
un moindre degré la phase II (glucuronoconjugaison). L’induc-
tion enzymatique par un médicament conduit à l’augmentation
du métabolisme de l’autre médicament, et donc à une baisse
d’efficacité (2, 3). Cette induction enzymatique résulte généra-
lement de l’augmentation de synthèse des différents CYP.
Parmi les inducteurs enzymatiques les plus couramment rencon-
trés, on peut citer le phénobarbital, la phénytoïne, la carbamazé-
pine, la rifampicine, la rifabutine, le méprobamate et la griséo-
fulvine.
La culture d’hépatocytes ou les suspensions de microsomes
humains permettent de déterminer la puissance inductrice des
médicaments.
Caractéristiques de l’induction enzymatique
Chronologie. Le délai de survenue et de disparition de l’effet induc-
teur est progressif et dépend de la demi-vie de l’inducteur. L’in-
duction peut être détectée au bout de
8 jours avec le phénobarbital, est
maximale au bout de 2 semaines et
persiste 2 à 3 semaines après l’arrêt
de l’inducteur (2). Avec la rifampi-
cine, l’induction atteint son maxi-
mum en 5 à 10 jours et disparaît en
5 à 10 jours suivant son arrêt (17).
Dose. L’induction enzymatique est
dose-dépendante : des doses éle-
vées de médicaments inducteurs
entraînent une majoration de l’in-
duction. Chez un patient, l’aug-
mentation de posologie du phéno-
barbital (100 mg/j à 200 mg/j)
diminuait les taux plasmatiques de warfarine (18).
En pathologie cardiovasculaire, certains médicaments voient leur
efficacité diminuée par augmentation de leur métabolisme lors
d’association avec des inducteurs enzymatiques. Parmi ceux qui
nécessitent une adaptation de posologie (augmentation de poso-
logie du médicament induit) et une surveillance clinique et/ou
biologique figurent la majorité des dihydropyridines, l’hydro-
quinidine (Sérécor®), la quinidine (Longacor®), le disopyramide
(Rythmodan®) et les antivitamines K.
CONCLUSION
En thérapeutique cardiovasculaire, seules certaines interactions
du type inhibition enzymatique sont essentielles à considérer.
La Lettre du Cardiologue - n° 325 - février 2000
25
DOSSIER

La Lettre du Cardiologue - n° 325 - février 2000
26
DOSSIER
L’administration d’un comprimé, prescrit seul ou avec des inhi-
biteurs enzymatiques, peut entraîner des concentrations plasma-
tiques différentes (normales ou décuplées) (19).
Afin d’évaluer le potentiel d’inhibition ou d’induction enzyma-
tique médié par les CYP, des modèles in vitro puis in vivo ont
permis de mettre en évidence des interactions médicamenteuses
potentielles.
Certaines interactions sont connues des patients et parfois même
utilisées : aux États-Unis, par exemple, nombre de patients hyper-
tendus ou greffés ont diminué par deux leur facture mensuelle de
félodipine ou de ciclosporine en prenant très régulièrement du
jus de pamplemousse. ■
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Guengerich F.P. Cytochrome P4503A4 and role in drug metabolism. Annual
Rev Pharmacol Toxicol 1999 ; 39 : 1-7.
2. Hansten and Horn’s. Drug interactions analysis and management.
3. Thummel K.E., Wilkinson G.R. In vitro and in vivo drug interactions involving
human CYP3A. Ann Rev Pharmacol Toxicol 1998 ; 38 : 389-430.
4. Martinowitz U. et coll. Interaction between warfarin sodium and amiodarone.
N Engl J Med 1981 ; 304 : 671.
5. Sakave H., Akamatsu K., Hirabayashi Y. Effects of prolonged oral cimetidine,
ranitidine and famotidine therapy on antipyrine elimination. Clin Ther 1987 ; 9
(6) : 602-6.
6. Cohen I.A. et coll. Cimatidine theophylline : interactions effects of age and
cimetidine dose. Ther Drug Monit 1985 ; 7 : 426.
7. Tetts et coll. Drug interactions with fluconazole. Med J Aust 1992 ; 156 : 365.
8. Tseng C.Y. et coll. Formation of morphine from codeine in chinese subjects of
different CYP2D6 genotypes. Clin Pharmacol Ther 1996 ; 60 : 177.
9. Lindholm A. et coll. Verapamil inhibits cyclosporin metabolism. Lancet 1987 ;
1:1262.
10. Pochet J.M. et coll. Cyclosporine-diltiazem interactions. Lancet 1986 ; 1 :
979.
11. Ahonen J et coll. Effect of diltiazem on midazolam and alfentanil disposition
in patients undergoing coronary artery bypass grafting. Anesthesiology 1996 ;
85 : 1246.
12. Trohman R.G. et coll. Increased quinidine plasma concentrations during
administration of verapamil : a new quinidine-verapamil interaction. Am J
Cardiol 1986 ; 57 : 706.
13. Curel P. Itraconazole and fluconazole interactions : similarities and diffe-
rences. Reactions 1998 ; 699 : 3.
14. Maury Ph., Vuille C., Metzger J. et coll. Intoxication sévère à l’acétate de flé-
caïnide. Arch Mal Cœur 1999 ; 92 (2) : 273-7.
15. Fossum Graham C., Turner W.M., Jones J.K. Lidocaine-propranolol interac-
tions. N Engl J Med 304 (21) : 1301.
16. Bedford T.A., Rowbotham D.J. Cisapride : drug interactions of clinical signi-
ficance. Drugs Safety 1996 ; 15 : 167-75.
17. Heimark L.D. et coll. The mechanism of the warfarin-rifampicin drug inter-
action in humans. Clin Pharmacol Ther 1987 ; 42 : 388.
18. Whitfield J.B. et coll. Changes in plasma gamma-glutamyl transpeptidase
activity associated with alterations in drug metabolism in man. Br Med J 1973 ;
1:316.
19. Jaillon B., Dupuis B., Dhan R. Études électrophysiologiques précliniques et
cliniques en vue de la prédiction d’un effet proarythmique (torsade de pointe)
iatrogène. Thérapie 1997 ; 52 : 271-80.
FMC
1. Citez deux exemples d’interactions par inhibition enzymatique qui conduisent à une contre-indication.
2. Conduite à tenir devant l’association inducteur enzymatique et hydroquinidine, quinidine, disopyramide et AVK.
3. Quel type d’interaction peut-il exister entre amiodarone et AVK ?
RÉPONSES
RÉPONSES
FMC
1. Statines et imidazolés antifongiques.
Cisapride et macrolide.
2. Il faut augmenter leur posologie, associée à une surveillance clinique et/ou biologique.
3. L’amiodarone (Cordarone®) est un agent inhibiteur du métabolisme enzymatique hépatique des anticoagulants oraux.
L’effet inhibiteur de l’amiodarone se manifeste en général après une semaine de traitement, atteint son niveau maxi-
mal après un mois et disparaît quelques mois après son arrêt, compte tenu de sa longue demi-vie.
AUTOQUESTIONNAIRE
AUTOQUESTIONNAIRE
1
/
4
100%