Landry BZD

FORMATION CONTINUE
Mise à jour sur les considérations pharmacocinétiques,
pharmacodynamiques et les interactions
médicamenteuses dans le choix d’une benzodiazépine
Update on the pharmacokinetics and
pharmacodynamic properties, drug interactions and
therapeutic usage of benzodiazepines
P. Landry
a,
*, M. Gervais
b
, K.P. O’Connor
c
a
Module de psychopharmacologie, centre de recherche Fernand-Seguin, clinique de psychopharmacologie, pavillon Bourget,
hôpital Louis-Hyppolite-Lafontaine, 7401, Hochelaga, H1N 3M5, Montréal, Québec, Canada
b
Hôpital Louis-Hyppolite-Lafontaine, Montréal, Québec, Canada
c
Centre de recherche Fernand-Seguin, université de Montréal, hôpital Louis-Hyppolite-Lafontaine, Montréal, Québec, Canada
Disponible sur Internet le 25 juillet 2008
Résumé
Au cours des 50 dernières années, nous avons cumulé beaucoup d’informations cliniques sur les aspects
pharmacocinétiques, pharmacologiques, les interactions médicamenteuses et l’utilisation thérapeutique des
benzodiazépines. L’objectif de cette revue de la littérature est d’identifier les principales différences entre les
benzodiazépines afin d’aider le prescripteur dans son choix de traitement. La demi-vie demeure l’une des
caractéristiques la plus importante dans le choix d’une benzodiazépine, celles ayant une demi-vie intermédiaire (six à
12 heures) étant recommandées pour un usage ponctuel ou pour pallier une insomnie initiale, alors que les
benzodiazépines avec une longue demi-vie (plus de 12 heures) sont recommandées lorsqu’un effet plus soutenu est
souhaité. Néanmoins, ces dernières comportent le risque d’augmenter l’occurrence d’effets secondaires dus à
l’accumulation du médicament. Leur métabolisme hépatique nous contraint à utiliser les benzodiazépines avec
précaution chez les patients souffrant d’une maladie du foie, chez les personnes âgées ou les personnes qui prennent
des médicaments modifiant l’activité enzymatique hépatique. Chez ces patients, il est préférable d’utiliser le lorazépam,
l’oxazépam ou le témazépam, puisqu’ils sont peu métabolisés par la voie hépatique. Pour plusieurs, mais pas pour toutes les
benzodiazépines, les risques d’interactions médicamenteuses concernent principalement les réactions d’oxydation
médiées par l’isoenzyme P-450 CYP3A4. Les antibiotiques kétoconazole et érythromycine, les corticostéroïdes,
certains antidépresseurs de la classe des inhibiteurs spécifiques du recaptage de la sérotonine (ISRS) et le jus de
pamplemousse sont tous des inhibiteurs du métabolisme médié par le cytochrome P-450 CYP3A4 et peuvent donc
provoquer une accumulation de benzodiazépines métabolisées par cette voie lorsqu’ils sont administrés conjointement.
Dans les situations qui exigent l’usage d’une benzodiazépine intramusculaire, le lorazépam est préféré à d’autres en raison
d’une absorption plus fiable et de son action rapide. Le mode d’action des benzodiazépines consiste à potentialiser l’effet
inhibiteur de l’acide gamma-aminobutyrique (GABA) dans le système nerveux central et, à ce niveau, il ne semble pas y
avoir de différence entre elles, sauf pour le clonazépam qui agit également sur la transmission sérotoninergique. Cette
propriété augmenterait l’effet anxiolytique du clonazépam et pourrait expliquer son efficacité à réduire les rechutes chez
certains déprimés. Le récepteur Gaba est constitué de cinq sous-unités protéiniques, elles-mêmes organisées sous la forme
d’une rosette. Les benzodiazépines potentialisent l’effet du GABA en se liant aux sous-unités a,bet gde la rosette. La
Annales Médico-Psychologiques 166 (2008) 585–594
* Auteur correspondant.
Adresse e-mail : [email protected] (P. Landry).
0003-4487/$ see front matter ß2008 Elsevier Masson SAS. Tous droits re
´serve
´s.
doi:10.1016/j.amp.2008.06.008

liaison d’une benzodiazépine à la sous-unité a
1
, localisée surtout dans le tronc cérébral, favorise la sédation alors que la
liaison avec la sous-unité a
2
, localisée dans le système limbique, est davantage associée à une réponse anxiolytique. L’usage
prolongé d’une benzodiazépine modifie l’expression génique et réduit la synthèse des ARN messagers codant pour les
sous-unités a
1
,a
2
,a
3
et, possiblement, b. Cette modification dans la composition du récepteur GABA est en fait une
autorégulation du récepteur et expliquerait le phénomène de tolérance aux benzodiazépines. Toutefois, les aspects
pharmacodynamiques sont très similaires pour toutes les benzodiazépines et le clinicien n’a pas à en tenir compte dans son
choix de traitement. Ainsi, les aspects pharmacocinétiques et les interactions médicamenteuses constituent les éléments
les plus significatifs à considérer dans le choix d’une benzodiazépine.
ß2008 Elsevier Masson SAS. Tous droits réservés.
Abstract
Over the past 50 years, significant clinical information has been obtained on numerous aspects of benzodiazepines,
notably on their pharmacokinetics and pharmacodynamic properties, drug interactions and therapeutic usage. The main
objective of this review is to determine which of these aspects a clinician should consider when choosing a
benzodiazepine. Benzodiazepines can be distinguished by their half-lives and those with an intermediate half-life
(6–12 h) are indicated for punctual usage and for initial insomnia while those with a longer half-life (over 12 h) are
indicated for clinical situations requiring sustained therapeutic treatment. Benzodiazepines with longer half-lives are
more at risk of causing side effects due to accumulation of the drug. Most, but not all, benzodiazepines are metabolized by
the liver and should be used carefully in patients known to have a hepatic disease, in geriatric patients or in patients taking
other medication modifying hepatic metabolism. In these cases, lorazepam, oxazepam or temazepam should be chosen
because they are less dependant on hepatic metabolism. Most but not all benzodiazepines are metabolized by the
intestinal and hepatic cytochrome P-450 3A4 system. A number of medication namely ketoconazole, erythromycin,
corticosteroids, fluoxetine, fluvoxamine, sertraline, calcium channel blockers and grapefruit juice may increase plasma
concentrations of some benzodiazepines by inhibiting P-450 3A4 cytochrome activity. Cigarette smoking has been
reported to significantly increase the clearance of alprazolam. The mechanism of action of benzodiazepines consists in
increasing the inhibitory effect of gamma-aminobutyrique acid (GABA) in the central nervous system. GABA receptor
contains five sub-units organised in the form of a rosette. The canal in the centre of the rosette permits the entrance of
chloride ions and the opening of the channel is modulated by GABA through changing the configuration of the proteins
constituting the rosette. Benzodiazepines alone have no inhibitory activity on neurones but will potentiate the effect of
GABA by binding to the sub-units a,band gof the rosette. Binding to the sub-unit a
1
localised mainly in the brain stem will
increase sedation while binding to the sub-unit a
2
in the limbic system will confer anxiolytic properties to
benzodiazepines. No differences among benzodiazepines have been reported concerning their binding to different
sub-units. However, clonazepam does appear to modify serotoninergic transmission and this pharmacodynamic
characteristic may increase clonazepam inhibitory properties and also stabilise mood in patient with unipolar
depression. Benzodiazepines prolonged usage will modify the DNA expression and decrease synthesis m-RNA
coding for sub-units a
1
,a
2
,a
3
and possibly b. These modifications in the composition of GABA receptors are
thought to be a mechanism of autoregulation of the receptor and could explain tolerance which is observed in
prolonged usage of benzodiazepines. Overall, pharmacokinetics properties and drug interaction remain the most
important characteristics when choosing a benzodiazepine while pharmacodynamic properties have little relevance
because differences are not significant within this class of medication.
ß2008 Elsevier Masson SAS. Tous droits réservés.
Mots clés : Benzodiazépine ; Interactions médicamenteuses ; Pharmacocinétique ; Pharmacodynamique
Keywords: Benzodiazepine; Drug Interaction; Pharmacodynamic; Pharmacokinetic
1. INTRODUCTION
Depuis la commercialisation du chlordiazépoxide
(Librium
1
) en 1960, suivi du diazépam (Valium
1
) en 1963,
on estime que plus de 3000 benzodiazépines ont été
synthétisées. Entre 35 et 50 seraient actuellement disponibles
sur le marché international [2,8,23]. Au cours des 50 dernières
années, nous avons cumulé beaucoup d’informations cliniques
sur les aspects pharmacocinétiques, pharmacodynamiques, les
interactions médicamenteuses et l’utilisation thérapeutique des
benzodiazépines autant en médecine physique qu’en psychiatrie
[11,29]. Malgré les inquiétudes liées à leur pharmacodépen-
dance, les benzodiazépines offrent une avenue thérapeutique
temporaire ou à plus long terme pour plusieurs patients
atteints d’un trouble psychiatrique. D’ailleurs, les benzodiazé-
pines demeurent un des trois choix privilégiés par les
psychiatres dans le traitement d’un trouble anxieux réfractaire
à un antidépresseur [43]. Une étude récente indique qu’au
Canada l’usage de benzodiazépines n’a pas diminué au cours de
la dernière décennie. Dans la population générale, la fréquence
d’utilisation se situerait entre 4,9 et 6,1 %, et atteindrait 11 %
chez les gens ayant un diagnostic psychiatrique [26]. Dans cet
article, nous proposons de revoir les caractéristiques
pharmacologiques des benzodiazépines et d’utiliser cette
information pour guider le prescripteur dans son choix de
traitement.
P. Landry et al. / Annales Médico-Psychologiques 166 (2008) 585–594586

2. CONSIDÉRATIONS
PHARMACOCINÉTIQUES DANS LE CHOIX
D’UNE BENZODIAZÉPINE
Les aspects pharmacocinétiques sont parmi les caractéristi-
ques les plus importantes dans le choix d’une benzodiazépine.
La pharmacocinétique s’intéresse au processus de transforma-
tion d’un médicament dans le corps à partir de son absorption
dans la circulation sanguine, ensuite sa distribution à son site
d’action et, finalement, sa biotransformation et son élimination
par le foie et les reins.
2.1. Forme d’administration
La majorité des benzodiazépines peuvent seulement être
administrées par voie orale, mais quelques-unes sont également
disponibles pour administration par voie intramusculaire
et/ou intraveineuse. Le choix de l’une ou l’autre des voies
d’administration relève de considérations cliniques, par
exemple, une urgence thérapeutique commandant un début
d’action rapide, l’incapacité pour un patient de prendre un
médicament par la bouche en raison de nausées importantes ou
d’une perte de conscience, notamment lors d’une crise
épileptique, la nécessité de prendre des doses quotidiennes
multiples, le refus de coopérer, etc.
Les préparations sublinguales (S/L) ont été développées avec
l’idée qu’elles agiraient plus rapidement que les formes orales
standard. On croyait, en effet, qu’elles seraient rapidement
absorbées à travers la muqueuse buccale, malgré une petite
surface d’absorption, et qu’en drainant directement dans la
veine cave supérieure, elles seraient vite acheminées vers leurs
organes cibles. Cependant, les études récentes concernant le
lorazépam et le clonazépam, seules benzodiazépines couram-
ment disponibles sous forme S/L, aucune accélération
significative de la vitesse d’absorption et, conséquemment,
du début d’action n’a pu être mise en évidence par comparaison
aux comprimés standards [16,37]. D’autres études ont tout de
même avancé le contraire [1].
Pour ce qui est de la voie intramusculaire, elle provoque
un pic plasmatique plus rapide et marqué que la voie orale en
raison d’une vitesse et d’un taux d’absorption accru. Il y a
donc un effet clinique plus soudain et intense et cet outil
thérapeutique s’avère très utile et sécuritaire en situation
d’urgence pour calmer rapidement l’agitation ou l’agressivité,
par exemple, chez des patients psychotiques ou en manie qui
manifestent des comportements violents. L’absorption est
cependant plus lente dans le muscle grand-fessier que dans le
deltoïde [10] en raison d’une plus forte proportion de
tissus graisseux faiblement perfusés dans cette partie de
l’anatomie [3]. Cela est encore plus marqué chez les femmes
et les personnes obèses. Pour plusieurs pays occidentaux,
quatre benzodiazépines sont disponibles sous forme intra-
musculaire, soit le midazolam, le lorazépam, le chlordiazé-
poxide et le diazépam. Le midazolam est presque
exclusivement réservé à la préanesthésie. Parmi les trois
autres, seul le lorazépam est absorbé de manière fiable et
rapide ; ce qui expliquerait que les médecins le préfèrent au
diazépam et au chlordiazépoxide pour les injections
intramusculaire [25].
L’usage intraveineuse des benzodiazépines, quoique plus
rare, permet à 100 % de la médication d’être distribuée,
puisqu’elle se retrouve d’emblée dans la circulation sanguine.
L’action commence en quelques secondes à peine, tout au plus
quelques minutes. En effet, une fois injectée, la benzodiazépine
est rapidement acheminée vers les organes les mieux perfusés,
y compris le cœur, le foie, les reins et le cerveau. Son caractère
lipophile lui assure une grande affinité pour le tissu cérébral, où
elle se distribue aisément en diffusant à travers les jonctions
serrées des cellules endothéliales des capillaires cérébraux. Le
midazolam et le diazépam pourraient d’ailleurs avoir un début
d’action plus rapide que le lorazépam, car ils sont davantage
liposolubles et traverseraient donc plus vite la barrière
hématoencéphalique [17,18]. Cette approche est habituelle-
ment réservée pour le traitement de crises convulsives (status
épilepticus) en anesthésie ou chez le patient en delirium.
L’administration d’une benzodiazépine par voie rectale
demeure exceptionnelle mais utile chez des patients aux prises
avec certains problèmes médicaux qui compliquent la prise d’un
médicament par voie orale ou encore chez ceux qui refusent de
prendre une médication. Au Canada, la seule benzodiazépine
disponible sous cette forme est le diazépam. L’absorption par la
muqueuse rectale serait cependant erratique [3]. Tout comme
la voie sublinguale, la voie rectale permet d’éviter en partie
l’effet de premier passage hépatique. Cela est avantageux
puisque, durant le premier passage hépatique, qui a lieu
lorsqu’un médicament est administré par la bouche, une
certaine quantité de médicament est métabolisée par le foie
avant même de se lier aux protéines plasmatiques pour voyager
jusqu’à son site d’action.
L’alprazolam est disponible sous une forme à libération lente
(Xanax TS
1
) qui permet de limiter la prise quotidienne de la
médication à une ou deux prises par jour plutôt que trois ou
quatre, comme c’est le cas avec l’alprazolam régulier. De plus, la
libération de la médication étant plus graduelle, la formulation à
libération lente assure une concentration constante du
médicament pendant plusieurs heures, ce qui évite des pics
ponctuels et associés à des effets indésirables. Il faut informer le
patient de s’abstenir de mastiquer ou de briser le comprimé,
puisque le médicament perdra ses propriétés à libération lente.
2.2. Absorption
Comme règle générale, plus la vitesse et le degré
d’absorption d’une benzodiazépine sont grands, plus la
molécule a un pic plasmatique élevé. Cela se traduit par des
effets thérapeutiques rapides mais aussi par des effets
indésirables qui risquent d’être plus marqués. Une plus grande
rapidité d’absorption risque également de favoriser davantage
une dépendance psychologique en raison d’un soulagement
thérapeutique plus rapide. L’absorption des benzodiazépines,
comme de tout autre médicament, est affectée par plusieurs
variables, parmi lesquelles la voie d’administration, le véhicule
pharmaceutique, les caractéristiques physicochimiques de la
molécule (liposolubilité, degré d’ionisation), la médication
P. Landry et al. / Annales Médico-Psychologiques 166 (2008) 585–594 587

conjointe, etc. Pour la plupart des benzodiazépines prises par la
bouche, l’effet apparaît 30 minutes à deux heures après leur
ingestion [10]. Le bromazépam, le clorazépate, le diazépam, le
flurazépam et le triazolam sont parmi les benzodiazépines ayant
un début d’action rapide et sont habituellement moins utilisées,
sauf pour des situations commandant un effet thérapeutique
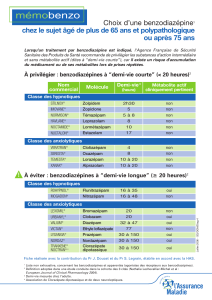
rapide, tel qu’une insomnie initiale (Tableau 1).
Dans leur forme orale standard, les benzodiazépines sont
toutes facilement absorbées par le tractus gastro-intestinal,
étant donné leur caractère liposoluble qui leur assure une
diffusion passive à travers l’épithélium de la muqueuse
intestinale vers les capillaires sanguins. La forme liquide est
absorbée plus rapidement que la forme solide, car elle n’a pas
besoin d’être dissoute par les sucs gastriques. Les comprimés
seraient également absorbés plus rapidement que les capsules.
Tout ralentissement de la vidange gastrique, par exemple par la
prise de nourriture, d’agents anticholinergiques ou antiacides,
entraîne par ricochet une absorption plus graduelle et, ainsi, un
pic plasmatique moins marqué [15]. L’effet ressenti par le
patient est donc moindre.
La quasi-totalité des benzodiazépines est dissoute par les
enzymes des sucs gastriques sans aucune modification
métabolique avant d’être absorbées à travers la muqueuse
intestinale. Seul le clorazépate subit une modification méta-
bolique dans l’estomac. Ce dernier est, en effet, un
promédicament qui est inactif dans sa forme originale. Il doit
d’abord être hydrolysé dans l’estomac en desméthyl-diazépam ;
ce qui permet alors à la molécule d’être absorbée et d’acquérir
des propriétés thérapeutiques. Toutefois, cette transformation
chimique ne retarde pas l’effet thérapeutique du médicament.
2.3. Distribution
La grande affinité des benzodiazépines pour les lipides
favorise non seulement leur distribution dans le cerveau mais
également leur redistribution vers les tissus adipeux péri-
phériques. Cela se traduit par une chute des concentrations
cérébrale et plasmatique en deçà de la concentration minimale
effective, et la molécule cesse alors de produire son effet
thérapeutique [9]. La phase de redistribution dans les tissus
adipeux périphériques a lieu d’autant plus vite que la
benzodiazépine a une liposolubilité élevée. Une plus grande
liposolubilité est davantage associée à l’apparition d’un trouble
mnésique, mais toutes les benzodiazépines auraient un impact
sur les capacités mnésiques, particulièrement la mémoire
épisodique [12]. Parmi les benzodiazépines, la liposolubilité est
la plus grande pour le diazépam, suivi du clorazépate, puis du
triazolam (Tableau 1).
2.4. Métabolisme, élimination et demi-vie et
implication clinique
Toutes les benzodiazépines sont hautement métabolisées
par le système enzymatique hépatique. Le foie doit préala-
blement les rendre hydrosolubles de manière à ce qu’elles
puissent être éliminées avec l’urine. Une partie des médica-
ments est également excrétée sous forme de bile et peut alors
être éliminée avec les selles ou encore être réabsorbée dans la
circulation pour subir un second passage hépatique avant d’être
excrétée dans l’urine.
Le flurazépam, le diazépam, le clorazépate et le chlordiazé-
poxide subissent une première réaction qui aboutit à la
formation de deux métabolites actifs ayant une longue demi-vie.
Par la suite, ces métabolites subissent une réaction d’oxydation
qui les transforme en dérivés 3-hydroxy. Ce sont aussi des
métabolites actifs. Un de ces métabolites, le nordazépam, est
métabolisé en oxazépam, une molécule avec une demi-vie
intermédiaire, elle-même, d’ailleurs, commercialisée. Finale-
ment, les dérivés 3-hydroxy sont conjugués à un acide
glucuronique et transformés en métabolites hydrosolubles
inactifs pouvant être excrétés dans l’urine.
Les benzodiazépines avec des métabolites actifs peuvent
s’accumuler et causer plus d’effets secondaires, notamment, au
niveau mnésique à moyen et long terme que les molécules sans
métabolites actifs. Ces benzodiazépines ne devraient donc pas
être utilisées chez les personnes âgées, car le risque
d’accumulation est d’autant plus grand qu’il y a diminution
de l’activité métabolique hépatique avec l’âge (Tableau 1).
Tableau 1
Caractéristiques pharmacocinétiques des benzodiazépines
Nom générique Début d’action Liposolubilité [39]
a
Demi-vie (heure) Métabolites actifs
Alprazolam 1–2 heures 0,54 9–20 Oui
Bromazépam 0,5–4 heures 0,24 8–30 Non
Chlordiazépoxide 1–4 heures – 4–29 Oui
Clonazépam 1–4 heures 0,28 19–60 Non
Clorazépate 0,5–2 heures 0,79 1,3–120 Oui
Diazépam 1–2 heures 1,00 30–200 Oui
Flurazépam 0,5–1 heures – 40–250 Oui
Lorazépam 1–6 per os
45–75 minutes intramusculaire
0,48 8–24 Non
Midazolam 0,5–1 minutes intraveineuse – 1–4 Oui
Nitrazépam 0,5–7 heures 0,29 15–48 Oui
Oxazépam 1–4 heures 0,45 3–25 Non
Témazépam 2,5 heures 0,50 3–25 Non
Triazolam 1–2 heures 0,64 1,5–5 Non
a
Un chiffre élevé indique une plus grande liposolubilité.
P. Landry et al. / Annales Médico-Psychologiques 166 (2008) 585–594588

Le triazolam, l’alprazolam et le midazolam sont également
transformés par oxydation en composés 3-hydroxy. Ces
métabolites actifs sont cependant très rapidement conjugués
par glucuronidation de sorte qu’ils ne s’accumulent pas de façon
significative. Malgré ces propriétés pharmacocinétiques avan-
tageuses, la courte demi-vie du triazolam est mise en cause
pour expliquer une amnésie antérograde et d’autres troubles
cognitifs liés à l’usage de ce médicament [22]. Soulignons que le
métabolisme hépatique du triazolam serait peu modifié avec
l’âge chez la femme, mais réduit chez l’homme [19].
Quant au lorazépam, à l’oxazépam et au témazépam, qui
sont eux-mêmes des dérivés 3-hydroxy, ils n’ont pas besoin de
subir une oxydation préalable et sont d’emblée conjugués à un
acide glucuronique. Leur élimination ne produit aucun
métabolite actif. L’altération de la fonction hépatique affecte
très peu leur élimination. Pour cette raison, ils représentent un
premier choix chez les personnes âgées et les patients souffrant
d’un trouble hépatique, tel qu’une cirrhose ou une hépatite.
Quant aux dérivés 7-nitro, comme le clonazépam, ils sont
métabolisés en amines inactives, ensuite acétylés et excrétés.
L’élimination des benzodiazépines et de leurs dérivés par
métabolisme hépatique et excrétion rénale se fait à un taux
propre à chaque molécule. Ce taux est représenté par le temps
de demi-vie d’élimination (t
1/2
). Les dérivés d’un même sous-
groupe ont des demi-vies d’élimination comparables, puisqu’ils
sont biotransformés par les mêmes voies métaboliques [8].
Ces temps de demi-vie servent à classer les benzodiazépines
en molécules avec durée d’action courte (t
1/2
inférieur à
cinq heures), intermédiaire (t
1/2
5–24 heures) ou longue
(t
1/2
supérieur à 24 heures) [20].
La demi-vie des métabolites actifs peut contribuer à la durée
d’action de la molécule mère. Le flurazépam en est un exemple
éloquent, puisque sa demi-vie est d’environ deux à trois heures
alors que la demi-vie de son métabolite principal, le N-
désalkylflurazépam, est de plus de 50 heures [2]. À cet égard, le
taux d’élimination du lorazépam, de l’oxazépam et du
témazépam est un index plus fiable de leur durée d’action
puisqu’ils ne produisent aucun métabolite actif [23].Ilenvade
même pour le triazolam et le midazolam, dont les métabolites
actifs sont très rapidement éliminés.
Cela étant dit, même si une benzodiazépine a une plus
longue demi-vie d’élimination, elle n’a pas nécessairement une
durée d’action plus longue. Par exemple, le diazépam a une plus
longue demi-vie que le lorazépam mais sa plus grande
liposolubilité réduit la durée de son action. Le diazépam est
donc plus promptement redistribué en périphérie que le
lorazépam ; ce qui met fin plus rapidement à son action sur ses
récepteurs centraux [10].
Avec un usage prolongé, en revanche, le diazépam
s’accumule et son effet est soutenu malgré sa grande
liposolubilité. Cela est également vrai pour les autres
benzodiazépines avec longue demi-vie d’élimination, par
exemple, le chlordiazépoxide ou le clonazépam. L’accumulation
est de plus favorisée par des doses rapprochées. Cette
accumulation se poursuit jusqu’à ce que la benzodiazépine
atteigne une concentration plasmatique à l’équilibre. La plupart
des médicaments, y compris les benzodiazépines et leurs
métabolites, y parviennent après cinq demi-vies. L’élimination
plasmatique d’un médicament nécessite également cinq demi-
vies. Quant aux benzodiazépines avec de plus courtes demi-
vies, elles s’accumulent moins, surtout si les intervalles entre les
doses sont importants.
L’accumulation est souhaitable lorsqu’on désire un effet
soutenu, par exemple pour traiter une symptomatologie
anxieuse chronique, des tics, une dystonie tardive ou une
épilepsie, raison pour laquelle on utilise alors les benzodiazé-
pines ayant une longue demi-vie. L’accumulation empêche les
fluctuations plasmatiques et la survenue de symptômes
interdoses. En revanche, l’accumulation est moins souhaitable
lorsqu’on recherche un effet ponctuel, par exemple pour
traiter une insomnie initiale. À cet égard, le choix d’une
benzodiazépine avec une demi-vie intermédiaire limitera l’effet
de nausée au lendemain de la prise de médicament. Pour ce qui
est des benzodiazépines ayant une courte demi-vie, elles ne
sont habituellement pas recommandées sur une base régulière
en raison d’un risque plus élevé d’effets rebonds et de troubles
mnésiques. Une benzodiazépine avec une longue demi-vie peut
présenter certains avantages en réduisant l’intensité des
symptômes de sevrage lorsqu’elle est cessée rapidement, mais
les caractéristiques pharmacocinétiques ont peu d’importance
lorsque la diminution se prolonge sur plusieurs semaines ou
plusieurs mois [40,42].
3. INTERACTIONS MÉDICAMENTEUSES DANS
LE CHOIX D’UNE BENZODIAZÉPINE
3.1. Interactions pharmacocinétiques
Par définition, une interaction pharmacocinétique produira
une modification du taux plasmatique d’un médicament. Ce
type d’interaction lié à l’usage de benzodiazépines peut
survenir à l’un ou l’autre des processus d’absorption, de
distribution, de métabolisme ou d’élimination. Ainsi, la
modification de l’acidité gastrique avec les antiacides peut
diminuer la vitesse d’absorption des benzodiazépines, notam-
ment du chlordiazépoxide et du diazépam. Par ailleurs, les
benzodiazépines et leurs métabolites se lient aux protéines
plasmatiques et toute condition qui réduit la disponibilité des
protéines peut augmenter la fraction libre de médicament dans
le plasma, soit la fraction libre active qui peut se lier à son
récepteur et produire un effet thérapeutique ou toxique. Entre
autres, l’âge avancé, la malnutrition et la cirrhose hépatique
sont des conditions caractérisées par une diminution de la
synthèse de protéines et peuvent donc être accompagnés
d’une fraction libre ou active augmentée du médicament.
L’administration conjointe de médicaments qui rivalisent pour
un même site de liaison aux protéines, par exemple l’acide
valproïque et le diazépam, peut également augmenter la
fraction libre de benzodiazépine plasmatique. Il semble
cependant qu’aucune conséquence clinique d’importance
n’ait pu être démontrée jusqu’à maintenant en lien avec ce
phénomène [8,16,23].
C’est au niveau du métabolisme des benzodiazépines par les
enzymes du cytochrome P450 (CYP450) que les interactions
P. Landry et al. / Annales Médico-Psychologiques 166 (2008) 585–594 589
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%