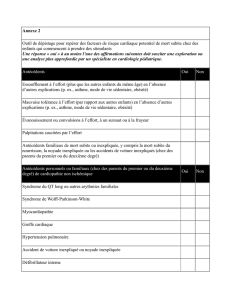
Pr Vincent Probst

DIU de rythmologie et stimulation cardiaque
Titre : Autres canalopathies : QT long, QT
court, Tachycardies ventriculaires
cathéconergiques
Orateur : Vincent PROBST
Le 27 janvier 2012

Nantes
Nantes
Long QT syndrome
Pr Vincent Probst,
Centre de référence pour la prise en charge des
maladies rythmiques héréditaire et USIC
L’institut du thorax, Nantes

Long QT syndromes
¾Rare disease 1/5 to 20000 people
¾Abnormal prolongation of the QTc>440 ms
¾Ventricular arrhythmias
¾Sudden death
¾Without cardiac morphological
abnormalities

Long QT syndromes
¾Clinically and genetically heterogeneous:
¾10 genes identified:
95 genes encoding for K channel
•
LQT1 and LQT5= KCNQ1 and KCNE1= Iks
•
LQT2 and LQT6= KCNH2 and KCNE2= Ikr
•
LQT7= Andersen syndrome = KCNJ2 (Kir
2.1)=IK1
93 genes inducing modification of the Na current
•
LQT3= SCN5A=INa
•
LQT9= Caveolin-3 gene
•
LQT10= SCN4A
91 gene for anchor protein
•
LQT4= Ankyrine
B
91 gene encoding for Ca++ channel
•
LQT8= CACNA1C= ICaL= Timothy syndrome

Three questions
¾How to perform the diagnosis?
¾How need treatment?
¾How to treat?
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
1
/
69
100%