G7-8, Dr V. Laugel Cours du 19/09/12 de 10h à 12h Master

G7-8 1/13
G7-8, Dr V. Laugel
Cours du 19/09/12 de 10h à 12h
Master physiopathologie Stéphane GIORGIUTTI et Geoffrey GINOT
_________________________________________________________________________________
Maladies de la réparation et de la transcription
INTRODUCTION
Les différentes maladies que nous allons étudier vont être classées en fonction de la lésion
sur l’ADN et selon les mécanismes de réparation comme nous pouvons le voir sur le schéma
ci-dessous. L’ADN est sans cesse agressé ce qui cause un nombre important de lésions par
jour dans chaque cellule. Si ces lésions s’accumulent sans mécanisme de réparation les
cellules en questions vont alors mourir ou devenir cancéreuses.
La réparation de ces lésions est fondamentale dans l’évolution des espèces.
Nous allons distinguer 4 grandes familles d’agents responsables de lésions :
1. Les rayons X/ Radicaux libres / Agents alkylants : ils vont oxyder la guanine,
transformer T en U ce qui va aboutir à une rupture simple brin et une perte de base.
Ces lésions sont simples à réparer, la double hélice est conservée, l’information
génétique est toujours présente. Elle est rapidement réparée par BER.
Plus le mécanisme de réparation est important moins il supporte d’altérations. Il est tellement
fondamental que s’il n’existe pas la cellule meurt donc il n’existe pas de maladie à
proprement parlé.
2. Les erreurs de réplication : ce sont des mismatch (A-G / T-C) où il se fait un mauvais
appariement, cette anomalie est directement due au fonctionnement de la cellule
(ADN polymérase et compagnie). Des enzymes vont reconnaitre cette formation et la
réparer, la non reconnaissance de ces anomalies entraine des cancers familiaux.

G7-8 2/13
3. Les rayons UV/ Les hydrocarbures aromatiques : UVA oxydatif ; UVBplus
énergétique, frappe l’ADN et crée une lésion de 2 bases adjacentes ; UVC crée
une liaison covalente. La dimérisation de base par liaison covalente modifie la forme
de la double hélice, nous aurons donc une distorsion de l’hélice qui va bloquer les
enzymes de la transcription (polymérase)
Le système de réparation NER va permettre la resynthèse du brin lésé. Il est donc
logique de trouver une hypersensibilité aux UV lors d’une anomalie de ce système de
réparation (Syndrome de Cockayne, Xeroderma pigmentosum, Trichothiodystrophie)
4. Les rayons X/ Les agents alkylants : la lésion va se produire sur les 2 brins en même
temps avec la formation d’une liaison covalente entre les deux brins de l’ADN. La
réparation nécessitera donc de casser les 2 brins de l’ADN puis de relier les 2 brins.
Ce mécanisme de réparation nécessite donc une recombinaison-religation qui va
induire des cross-over, des cassures chromosomiques. (ataxie-télangiectasie,
anémie de Fanconi, Syndrome de Bloom, Syndrome de Werner
I. MALADIES DE LA NER/ TRANSCRIPTION
Ici, un rayon UV va frapper l’ADN, donnant une laison covalente entre 2 bases
adjacentes, bloquant la fonction de la cellule.
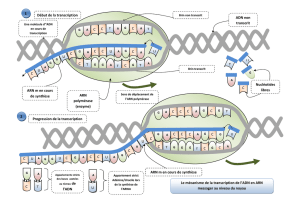
Mécanisme rapide de réparation : c’est le TC-NER (transcription couple NER). Comme
nous le savons il n’y a qu’une partie de l’ADN qui est transcrite, si l’anomalie se trouve au
niveau d’une transcription active de gène, la machinerie transcriptionnelle va se bloquer
et bloquer la fonction de la cellule. La RNA polymérase II est bloquée par la lésion et ceci
va déclencher le mécanisme de réparation. La RNApolII est toujours accompagnée de
deux hélicases (XPB et XPD) qui vont ouvrir la double hélice pour permettre la
transcription. CSA et CSB sont présentes lorsqu’il y a des blocages et vont reculer ou
enlever la polymérase pour appeler les enzymes qui coupent le brin abimé.
C’est un mécanisme de sauvetage immédiat, lorsque la transcription est bloquée
Mécanisme lent : ce mécanisme est plus rigoureux et systématique, c’est le GG-NER
(global genom NER), il se fait par screening de la double hélice.
- XPC screen la double hélice et se lie aux anomalies de celle-ci, il a une affinité pour la

G7-8 3/13
double hélice déformée. C’est une sorte de drapeau signalant l’anomalie.
- XPC va appeler XPE et XPA qui sont des protéines de recrutement qui elles vont
recruter XPG et XPF qui vont couper l’ADN (coupure 5’ et 3’).
- Suppression de 20 nucléotides sur l’un des brins, excision, il suffit maintenant de
resynthétiser en fonction du brin matrice et de lier le tout.
Le problème de ce mécanisme est sa lenteur
Ensuite la voie de réparation est commune aux deux voies de réparation (cf schéma)
Histoire : les protéines portent leur nom XP et CS car les maladies ont permis de découvrir
l’existence de ces protéines.
Test de réparation par irradiation de fibroblastes :
La GG-NER va être testée par l’UDS, elle consiste en l’incorporation de thymidine tritiée,
les cellules vont donc l’utiliser uniquement si elles se multiplient. Si l’on met des UV sur ces
cellules, il y a toujours des cellules en réplication, qui vont fixer beaucoup de thymidine tritiée
et d’autres qui n’étaient pas en réplication et qui vont quand même incorporer un peu de
thymidine tritiée parce qu’elles réparaient leur ADN. Ici on va donc regarder la synthèse
d’ADN en dehors de la phase S, donc la quantité de thymidine tritiée incorporée après
irradiation UV est proportionnelle à l’activité de réparation de la cellule. Si la cellule
n’incopore pas du tout de thymidine tritiée alors que la cellule a plein de lésion sur son ADN
c’est que la cellule ne répare pas.
La TC-NER va être testée par la RRS, elle consiste en l’incorporation d’uridine tritiée et on
va regarder comment la cellule relance sa transcription après l’avoir arrêté à cause des
lésions (les lésions UV bloquaient la transcription, le fibroblaste se fige). Lors du relancement
de la machine de transcription, le fibroblaste va incorporer de l’uridine tritiée, les cellules
incapables de relancer la transcription ne fixeront plus d’uridine.

G7-8 4/13
A. Xeroderma pigmentosum (XP)
Sa première description a été faite en 1870 par Kaposi (le même que le sarcome). C’est une
maladie à transmission autosomique récessive. Elle est rare (1/250 000 en Europe ; 1/40
000 en Afrique du Nord et au Japon). Il existe plusieurs groupes de complémentation :
plusieurs malades avaient cette maladie et ils ont fusionné les maladies deux à deux, si deux
cellules de malades différents, une fois fusionnées donnaient une cellule normale, cela
voulait dire que la mutation dans les 2 protéines n’étaient pas les même : au moins 8 gènes
connus : XPA à XPG + XPV (il faut rajouter de la caféine dans la manipulation d’avant pour
activer les processus chez les XPVariant). Les protéines touchées sont des protéines de la
voie NER (sauf pour XPV).
Clinique : Dermato :
- Peau atrophique, sèche, parcheminée (xeroderma = peau sèche)
- Anomalie de la pigmentation (pigmentosum)
- Photosensibilité, photophobie
- Susceptibilité aux cancers cutanés dans les zones exposées au soleil (+ cancers viscéraux)
car certaines cellules ne sont pas détruites mais vont être réparées mais laissant une
anomalie ce qui fait que ces patients ont un risque 1000 fois supérieur à la moyenne de
développer un cancer cutané. Pour ce qui est des cancers viscéraux, ceci est plus
surprenant, mais ils ont plus de risque de cancer du colon, de l’ovaire… d’autres agents que
les UV peuvent éventuellement provoquer les même lésions de l’ADN ce qui expliquerait
qu’il y a des cancers viscéraux.
On appelle ces patients « les enfants de la lune » car le jour ils ne peuvent pas sortir sans
être touché par les UV (même quand il pleut !) et doivent sortir totalement couvert. Il existe
des associations de familles qui organisent des soirées/week end où tout se passe la nuit,
cependant il faut faire attention à certaines lampes qui vont émettre des UV.
UDS
0
10
20
30
40
50
60
70
80
331 64 73
Dose UVC J/m2
Grains par noyau
Témoin normal
Témoin XP C
Trichothio-
dystrophie XPD
Témoin
Cockayne
UDS
0
10
20
30
40
50
60
70
80
331 64 73
Dose UVC J/m2
Grains par noyau
Témoin normal
Témoin XP C
Trichothio-
dystrophie XPD
Témoin
Cockayne

G7-8 5/13
Le problème est de détecter la
maladie assez tôt pour éviter le cancer, souvent la pathologie n’est découverte que vers
5ans et le cancer est déjà présent.
Clinique : Neuro :
Ce qui est surprenant, c’est que dans 20 à 30% des cas de XP nous trouvons un retard
mental. Il est probable que dans le métabolisme neuronal il y a une accumulation de lésions
qui ne sont pas réparés et provoque une apoptose des neurones.
A l’extrême ils ont une microcéphalie, un retard de croissance, un retard mental sévère, les
signes dermato du XP, c’est le syndrome De Sanctis-Cacchione, qui est une forme sévère et
rare de XP.
Biologie :
Le diagnostic se fait par biopsie cutanée pour confirmer la clinique, on irradie les fibroblastes
avec des UV et l’on constate que UDS et RRS sont diminuées sauf le patient muté dans
XPC où RSS est normale et les patients XPV où RRS et UDS sont normales. Les patients
XPC n’ont jamais de retard mental.
B. Syndrome de Cockayne / Syndrome COFS
Sa première description date de 1936 par E. Cockayne sur un frère et une soeur, ces
enfants ont un retard de croissance, un retard mental, une photosensibilité, une rétinopathie
pigmentaire progressive, une surdité de perception, des anomalies dentaires, une
dysmorphie particulière (en bec d’oiseau).
On reconnait la photosensibilité mais la première bizarrerie est qu’ils sont beaucoup moins
sensibles que les XP, ils ont des coups de soleil mais n’ont jamais de cancer. Il doit y avoir
d’autres anomalies qui devraient être réparées par la même voie qui ne le sont pas, ou bien
On peut voir des mélanomes ainsi
que des lésions de la rétine. Ces
lésions ne touchent que des parties
exposées.
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%