Trsiomie 21

Syndrome de l’X fragile
(syndrome de Martin-Bell)
Épidémiologie
Le syndrome de l’X fragile est la première cause de déficience intellectuelle héréditaire
avec une fréquence de l'ordre de 1 sur 4000 chez les hommes et 1/7000 chez les femmes.
Le syndrome de l’X fragile est une maladie à transmission dominante liée au chromosome
X (transmission verticale et pas de transmission père fils) avec quelques particularités
(hommes vecteurs, expression incomplète chez la femme, augmentation du nombre de cas au
fur et à mesure des générations (paradoxe de Sherman)).
Diagnostic postnatal
Les signes cliniques ont été initialement rapportés par Martin Bell : triade déficience
intellectuelle, dysmorphie faciale et macroorchidie (post pubertaire).
Actuellement le diagnostic est souvent fait dans l’enfance chez des garçons présentant une
déficience intellectuelle légère à modérée (retard de langage) associée à des troubles du
spectre autistique et une hyperactivité.
Diagnostic biologique
Initialement (1977) un site fragile au niveau des bras longs d’un chromosome X, en Xq27.3,
dans des conditions particulières de culture, a été émis en évidence sur le caryotype.
Découverte du mécanisme moléculaire en 1991 : expansion de triplets CGG en 5’ UTR du
gène FMR1 au locus FRAXA. Le nombre de triplets est variable d’un individu à l’autre. En
fonction de nombre de triplets, on définit différents allèles :
- normal : entre 6 et 49 répétitions. Transmission stable du nombre de triplets, pas de
phénotype
- intermédiaire : entre 50 et 54 répétitions : instabilité possible lors de la méiose. Pas
de phénotype
- prémutés : entre 55 à 200 répétitions : les hommes transmettent leur prémutation à
toute leur fille de manière stable. Les femmes transmettent l’allèle prémuté dans 50%
des cas avec augmentation du nombre de triplets. Le risque de transition vers une
mutation complète augmente avec la taille de la prémutation et est supérieur à 99%
lorsque la prémutation maternelle comporte plus de 100 répétitions CGG
- mutés : > 200 répétitions : méthylation du gène FMR1, absence de protéine.
Expression de la maladie chez les garçons et chez environ 50 % des filles.
Dans de rares cas (< 1%), le syndrome est causé par une mutation ponctuelle ou une délétion
du gène FMR1.
En cas de suspicion de syndrome de l’X fragile il est recherché une expansion de triplets CGG
et une anomalie de méthylation du gène FMR1, par PCR, méthyl-PCR et ou Southern blot
avec ou sans étude la méthylation.
En pratique, le médecin doit informer le patient et ses parents (si enfant) de l’analyse
génétique qui sera réalisée, recueillir le consentement signé et faire prélever un tube sur
EDTA pour extraction d’ADN et recherche d’une expansion de triplets.

Description de nouveaux phénotypes
La compréhension des mécanismes moléculaires et le suivi médical des familles ont permis de
montrer, contrairement à ce qui était décrit il y a quelques années, que les patients porteurs
d’un allèle prémutés ne sont pas toujours sains.
20 % des femmes prémutées ont un taux élevé de FSH, une infertilité et un risque de
ménopause précoce (avant l’âge de 40 ans) : syndrome FXPOI, contre 1% dans la
population générale. Une prémutation du gène FMR1 peut donc être recherchée devant tout
signe d’insuffisance ovarienne inexpliqué.
Les hommes (et parfois les femmes) porteurs de la prémutation peuvent développer des
signes neurologiques d’apparition tardive : ataxie cérébelleuse, tremblement intentionnel,
signes pyramidaux et parfois déclin intellectuel : syndrome FXTAS. La pénétrance de
l’ataxie et des tremblements est de 17%, 38%, 47% et 75% pour des hommes âgés de 50-59,
60-69, 70-79 et plus de 80 ans respectivement. La pénétrance est trois fois moins élevée pour
les femmes. Il y a une corrélation entre le nombre de répétitions et les symptômes moteurs et
cognitifs.
La prise en charge
Pas de traitement curatif.
L’éducation précoce et la qualité de la prise en charge initiale de l’enfant et des parents
tendent à diminuer le retard et augmenter l’autonomie.
La prise en charge doit être médicale, paramédicale, sociale et éducative, donc globale et
précoce
Place importante des associations : par exemple Goeland
Traitement
Dans le cadre du syndrome de l'X fragile, l'absence de la protéine FMRP crée une activité trop
importante des récepteurs mGluR5. Une molécule visant à bloquer cette voie glutamatergique
a été développée et testée sur une première cohorte de patients. Les résultats de cette étude
préliminaire ont montré un réel bénéfice sur les troubles du comportement sur un sous-groupe
particulier de patient présentant une méthylation complète. Des essais cliniques
complémentaires sont en cours.
Diagnostic prénatal (DPN)
Possibilité de proposer un DPN dans les cas suivants :
- femme porteuse d’une prémutation ou d’une mutation complète
- femme porteuse d’une délétion ou d’une mutation ponctuelle du gène FMR1
- très rarement : homme porteur d’une mutation complète.
La connaissance du sexe fœtal est indispensable à l’interprétation des données biologiques.
L’interruption médicale de grossesse ne sera envisagée que si le fœtus est porteur d’une
mutation complète.

Généralement le DPN est réalisé sur villosités choriales mais dans certains cas une ponction
de liquide amniotique est nécessaire, en particulier pour connaître le niveau de méthyation du
gène FMR1.
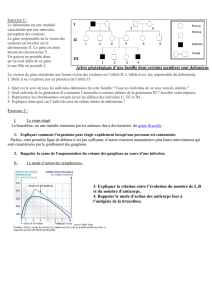
Conseil génétique
Il est complexe, car le diagnostic chez le cas index conduite à mettre en évidence d’autres cas
dans la famille, identifier les femmes conductrices (prémutées ou mutées), voir à faire des
diagnostics présymptomatiques chez des personnes prémutées. En effet, la détermination du
statut génétique chez les grands-parents peut générer un sentiment de culpabilité avec, en plus,
valeur prédictive du test en ce qui concerne le risque de FXTAS. L’information transmise lors
de la consultation de conseil génétique est donc très importante.
Une femme prémutée ou mutée à 50 % de risque de transmettre son allèle morbide à un de
ses enfants. Tous les garçons mutés seront malades. Cinquante pour cent des filles mutées
filles présenteront des symptômes, ce qui complique les décisions à prendre en période
anténatale. Devant ces difficultés certains couples souhaitent bénéficier d’un diagnostic pré
implantatoire.

FICHE FLASH
EPIDEMIOLOGIE
• Cause la plus fréquente de retard mental héréditaire.
• Touche un garçon sur 4000 et une fille sur 7000.
CLINIQUE
• Triade diagnostique :
Retard mental
Dysmorphie faciale
Macro-orchidie (post pubertaire)
mais phénotype très variable.
• Phénotype chez les personnes prémutées
femmes : syndrome FXPOI : ménopaux précoce
Hommes > femmes : syndrome FXTAS : ataxie, tremblements
GENETIQUE
• Transmission dominante liée à l'X.
• Expansion de triplet CGG en 5’UTR du gène FMR1 (Xq27.3)
• Mécanisme : allèle normal prémuté dans un spermatozoïde. Cet allèle est transmis de
façon stable (méiose paternelle) ou subi un phénomène d'expansion (augmentation du nombre
de CGG) au cours de la méiose maternelle.
• En fonction de la taille de l’expansion, on distingue les allèles suivants
normal : entre 6 et 49 répétitions.
intermédiaire : entre 50 et 54 répétitions : instabilité possible lors de la méiose.
prémutés : entre 55 à 200 répétitions : instabilité lors de la méiose maternelle
mutés : > 200 répétitions : méthylation du gène FMR1, absence de protéine. Expression de
la maladie chez les garçons et chez environ 50 % des filles.
• Le conseil génétique : basé sur
Le diagnostic des femmes conductrices
L’importance de la prémutation
DIAGNOSTIC
• Généalogie évocatrice + clinique.
• Cytogénétique : Site fragile sur le bras court du chromosome X.
• Biologie moléculaire : taille de l’allèle augmenté + méthylation anormale
DIAGNOSTIC PRENATAL
• Proposé aux femmes ayant eu un enfant atteint ou porteuse d’une mutation complète
• Réalisé à 10-11 SA à partir de villosités choriales
caryotype fœtal : sexe du fœtus.
Etude des allèles en biologie moléculaire
possibilité de diagnostic pré implentatoire
1
/
4
100%