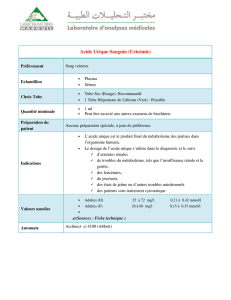
unité 14 la physiopathologie des troubles du metabolisme proteique

1
CURS5
Annéeuniversitaire2014-2015
UNITÉ14
LAPHYSIOPATHOLOGIEDESTROUBLESDUMETABOLISME
PROTEIQUE
Plandecours
I.Lestroublesdumétabolismedesprotéinesplasmatiques
1.Hypoprotéinémies
2.Hyperprotéinémies
3.Dysprotéinémies
II.Lestroublesdumétabolismedesaminoacides
III.Lestroublesdumétabolismedespurines
I.Lestroublesdumétabolismedesprotéinesplasmatiques
laprotéinémietotalelaconcentrationdesprotéinesplasmatiques
lesfractionsprotéiquesprincipalessontlessuivantes:
albumines(60%)
globulines(40%):
1-globulines(3,56,5%)
2-globulines(6-12%)
globulines(9-15%)
globulines(15-21%)
Lasynthèsedesprotéinesplasmatiques(15/jour)esteffectuéeauniveau:
Dufoiealbumine,globulines(80%),lesfacteursdecoagulation
Desplasmocyteslesimmunoglobulines
Desreinsrénine,érythropoïétine
Del’intestinapoprotéinesdequelquesfractionslipoprotéiques
Desglandesendocrinesleshormonespolypeptidiques
1.Hypoprotéinémieladiminutiondelaconcentrationdesprotéinesplasmatiques
Causesd’hypoprotéinémierelative:
hémodilution:
lesperfusionsmassives
larétentionhydrosaline (insuffisancerénaleaigue,insuffisancecardiaque
congestive)
Causesd’hypoprotéinémieabsolue:
Lapertedesprotéines
parvoierénale:syndromenéphrotique
parvoiedigestive:entéropathieexsudative
auniveaucutanépertedeplasmadanslesbrûlures
auniveaudel’appareilrespiratoire:bronchiectasies
delasynthèsedesprotéines
primaire(génétiquementdéterminée)
secondairedueunecarenceenaminoacides: inanition,malabsorption
secondairedueunemaladiedufoiel'hépatite
ducatabolismeprotéique
duniveaudeshormonescatabolisantes:diabètesucré,infections,tumeurs
DEPARTEMENTDESSCIENCESFONCTIONNELLES
PHYSIOPATHOLOGIE
RueTudorVladimirescu,14
300173Timisoara,
Tel/Fax:+40256493085

2
2.Hyperprotéinémiedelaconcentrationdesprotéinesplasmatiques
Causesd’hyperprotéinémierelative:
desétatsaccompagnésparhémoconcentration
ladéplétionhydrosaline (transpirationsprofuses,vomissementssévères,diarrhée,
diabèteinsipide,diabètesucré,etc.)
del’hématocrite
Causesd’hyperprotéinémieabsolue:
delaproductiond'immunoglobulinesdans:
infections
maladiesauto-immunes
plasmocytome(myélomemultiple),lamaladiedeWaldenström para
protéinémie et paraprotéinurie
Conséquences:
delapressiononcotiqueattractiondel'eaudel’intersticedanslelitvasculairerarement
hyper-volémie
delaviscositéduplasma
delasynthèsedesautresfractionsprotéiquestroublesdetransport,decoagulation,
immunologiques
3.Dysprotéinémie lechangementdurapportentrelesfractionsprotéiquesavecaltérationdu
rapportalbuminesglobulines
Typesdesdysprotéinémie:
a.Dysprotéinémiedel'inflammationaiguë
Causes:
lésionstissulaires(l’infarctusdumyocarde,lestumeurs,lesinterventionschirurgicales,les
infections,lescollagénases)déclenchementdelaréactiondephaseaiguëlasynthèse
desprotéinesdephaseaiguë(1-antitrypsine,1-anti-chimio-trypsine,1-orosomucoïde,2-
haptoglobine,2-céruloplasmine,protéineréactive,fibrinogène,lescomposantsdu
complément)
Caractéristiques:
des1-et2-globulines
desalbumines
b.Dysprotéinémiedel'inflammationchronique
Estidentiquel'inflammationaiguëdes-globulines(immunoglobulines)
c.Ladysprotéinémiedesmaladieshépatiques
Causes:hépatitechroniqueactive,cirrhosedufoieavec:
déficitdesynthèsedesalbuminesdûlalésiondeshépatocytes
delaproductiond'Igduel’hyperréactivitédutissuréticulo-endothélialdufoie
défautdedégradationhépatiqued'Ig
Caractéristiques:
desalbumines
deset-globulines
S’associeunehypoprotéinémie
l’aspectcaractéristiqued’ELFO:apparaîtle"dômecirrhotique"–parlafusiondessommets
et
d.Dysprotéinémiedusyndromenéphrotique
Causes:
glomérulonéphritesdelaperméabilitédufiltreglomérulaireprotéinurie3,5g/24h

3
Caractéristiques:
desalbuminesetdes-globulines(perteurinairedéficitd’augmentationcompensatoire
delasynthèsehépatiquedesalbumines)
2-et-globulines(compensatoiredeleursynthèsehépatique)
Elleestassociéel’hypoprotéinémie
II.Lestroublesdumétabolismedesaminoacides(AA)
1.LestroublesdeladégradationetdustockagedesAA
Laphenylcetonurie
L`alcaptonurie
L`albinisme
2.LestroublesdutransportdesAA
Lacystinurie
LamaladiedeHartnup
1.Lestroublesdeladégradationetdustockage
a.Laphénylcétonurie(l’oligophreniephenyl-pyruvique
Définition:untroublemétaboliquedéterminégénétiquementcaractérisépar:
delaphénylalaninesérique
del’éliminationparvoieurinairedelaphénylalaninelesproduitsdecatabolisme:
acidephénylpyruvique
acidephényl-lactique
acidephényl-acétique
toxiquespourleSNretardmentalsévère
Pathogenèse:
L'hyperphénylalaninémieestlerésultatde:
del`activitédelaphénylalaninehydroxylasequicatalyselaconversiondela
phénylalaninetyrosinedanslefoieetlesreins(laformeclassique)
Ledéficitendihydroptéridine-réductasequicatalyselasynthèseducofacteurdela
phénylalaninehydroxylasetétrahydrobioptérine(ptéridineréduite)
Manifestations:
Lésionscérébrales
retardmentalsévèreparla:
DiminutiondelaconcentrationdesAA(aminoacides)nécessaireslasynthèsedes
protéines
Diminutiondelasynthèsedelamyéline
Diminutiondelasynthèsedelanorépinephrineetdelasérotonine
Diminutiondelasynthèsedelamélanine
delaconcentrationplasmatiquedelaphénylalanine20mg/dldanslescasnontraités
del’éliminationurinairedescatabolitesdelaphénylalanine
desconcentrationsplasmatiquesd'autresacidesaminés
Diagnosticpositif:testd’inhibitionbactériennedeGUTHRIE
b.L`alcaptonurie
Définition:troublemétaboliqueprovoquéeparundéficitenhomogentysatedioxygénase,uneenzyme
impliquéedanslemétabolismedelatyrosineacidehomogentisiqueacidemaleil-acétique
Pathogenèse:déficitenzymatiqueentraîne:
l'accumulationdel'acidehomogentisique:
danslestissus(foie,reins)
danslesfluidesbiologiques(concentrationsériquefaible)éliminationrénale
rapide
Sonfixationaucollagènearticulaireochronose

4
L’ochronosedéterminel'arthritedégénérativeavecdesblessuresducartilagepar:
Irritationchimiquedirecte
L'inhibitiondessystèmesenzymatiquesimpliquésdanslemétabolismeducollagène
c.L`albinisme
Définition:défautgénétiqueauniveaudesmélanocytesmélanogenèsedéficitaire
Caractéristiques:
Dépigmentationdelapeaukératoseprécancéreuse
Dépigmentationoculairephotophobieetladiminutiondel'acuitévisuelle
2.LestroublesdutransportdesAA
a.Lacystinurie
Définition:troubleavectransmissionautosomiquerécessive,caractériséparuneaugmentationde
l'excrétionurinairedesAAdibasiques(Cystéine,Ornithine,Lysine,Arginine),dueladiminutionde
leursréabsorptiontubulairerénaleintestinale
Manifestations:
Sontdéterminéesparl’augmentationdel'excrétiondelacystéinecalculose:
rénale
urétérale
vésicale
b.LamaladiedeHartnup
Définition:carencedetransportrénaletintestinaldesAAmonoamine-mono-carboxyliques(ALA,
PHAL,LEU,ILEU,VAL,TRY,TRN)
Manifestations:
aminoaciduriesévère(5-10V.N.)
carenceentryptophanediminutiondelasynthèsedelaNIACINEetduNADPHsyndrome
cliniquedepellagre:
Dermatite
Diarrhée
III.Lestroublesdumétabolismedespurines
a.L’hyperuricémiemétabolique
b.L’hyperuricémierénale
c.Lagoutte
Lesnucléotidespuriniques(AMP,GMP,IMP)proviennentdetroissources:
Apportexogène
Turnovernormald’acidesnucléiquesaveclaréutilisationdesbasespuriniques("lavoiedu
sauvetage")
Labiosynthèse‘’denovo’’delaribose-5-P
Leproduitfinalducatabolismel'acideurique
a.L`hyperuricémiemétabolique
Définition:augmentationdelaproductiond'acideurique
Pathogenèse:
Activationdelasynthèsedenovodesnucléotidespuriniques
Diminutiondelaréutilisationdesbasespuriniques
L’augmentationduturnoverdesacidesnucléiquesdéterminel’hyperuricémiesecondaire:
lestroublesmyéloprolifératifsetlymphoprolifératifs
lesanémiehémolytiques
lespolyglobulies

5
b.L’hyperuricémierénale
Définition:diminutiondel'excrétionrénaledel'acideurique
Pathogenèse:
delafiltrationglomérulairedel’acideuriquedans:
lesaffectionsrénales:lesreinspolykystiques
deFGréabsorptiontubulairedansladéplétiondevolumelorsde:
ladiarrhée,transpiration,vomissements
l`insuffisancedelaCSR(carenceenALDO)
lediabèteinsipidenéphrogène
letraitementavecdiurétiques(!)
delasécrétiontubulairerénaledel'acideuriqueparl’inhibitioncompétitivedel'excrétion
del'acideuriquedansl’hyperproductiondesacidesorganiques:
l'acidocétosediabétique
l’acidoselactique
c.Lagoutte
Définition:troublecliniquemanifestépar
Hyperuricémieuricosurieresponsablesdela
néphropathieurique
lithiaseurique
Arthriteinflammatoireaiguërécurrenteparuneaccumulationaccruedecristaux
d'uratesauniveaudusynovialeetcartilagearticulaireaveclaformationdutophusgouteux
Pathogenèse:
Lesaccèsdegouttedéclenchésparlalibérationdescristauxdansl'espacearticulairesontproduitspar
lesmécanismessuivants:
laphagocytosedescristauxdesuratespardesleucocytesprovoquelarupturedes
membraneslysosomaleslibérationdesprotéaseslysosomalesdansleliquidesynovial
libérationdesmédiateursinflammatoires
l’activationsecondairedusystèmedeskininesetducomplémentinflammationaiguëlocale
auto-limitativeavecdestructionsminimes
Lescrisesdouloureusescommencentgénéralementauniveaudel'orteiletsontprécipitéspar:
l’alimentationricheenprotéines
lesdéshydratations
lesurchargemécaniquetraumad’articulations
1
/
5
100%