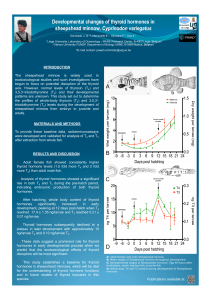
Universidade Federal do Rio Grande Do Sul Faculdade de Medicina

Universidade Federal do Rio Grande Do Sul
Faculdade de Medicina
Programa de Pós-Graduação em Ciências Médicas: Endocrinologia
Mírian Romitti
Metabolismo dos Hormônios Tireoidianos no Carcinoma Papilar de
Tireoide: Implicações na Tumorigênese e Crescimento Neoplásico
Porto Alegre
2014

Mírian Romitti
Metabolismo dos Hormônios Tireoidianos no Carcinoma Papilar de
Tireoide: Implicações na Tumorigênese e Crescimento Neoplásico
Tese de Doutorado apresentada ao Programa
de Pós-Graduação em Ciências Médicas:
Endocrinologia da Universidade Federal do
Rio Grande do Sul como requisito parcial
para obtenção do título de Doutor em
endocrinologia.
Orientadora: Profa. Dra. Ana Luiza Maia
Porto Alegre
2014

CIP - Catalogação na Publicação
Elaborada pelo Sistema de Geração Automática de Ficha Catalográfica da UFRGS com os
dados fornecidos pelo(a) autor(a).
Romitti, Mírian
Metabolismo dos Hormônios Tireoidianos no
Carcinoma Papilar de Tireoide: Implicações na
Tumorigênese e Crescimento Neoplásico / Mírian
Romitti. -- 2014.
59 f.
Orientadora: Ana Luiza Silva Maia.
Tese (Doutorado) -- Universidade Federal do Rio
Grande do Sul, Faculdade de Medicina, Programa de Pós-
Graduação em Ciências Médicas: Endocrinologia, Porto
Alegre, BR-RS, 2014.
1. Carcinoma Papilar de Tireoide. 2. Hormônios
tireoidianos. 3. Desiodase tipo 3. I. Silva Maia,
Ana Luiza, orient. II. Título.

2
AGRADECIMENTOS
À Profa. Dra. Ana Luiza Maia, a grande responsável por este trabalho, os meus
agradecimentos pela orientação, pelas inúmeras oportunidades, por toda a confiança,
paciência e amizade. Obrigada por ter apostado em mim e contribuído de maneira
extraordinária para a formação da pessoa que sou hoje.
Aos colegas e muito mais do que isso, amigos do Grupo de Tireoide, Lucieli Ceolin,
Carla Vaz Ferreira, Simone Wajner, Rafaela Vanin Pinto Ribeiro, Helena Cecin Rohenkohl,
Carla Krause, Shana Weber, Juliano Dalla Costa, Clarissa Capp, Leonardo Leiria, Debora
Rodrigues Siqueira, Nadja Zennig, Rafael Selbach Scheffel e José Miguel Dora pela preciosa
amizade e por compartilharem tantos momentos agradáveis no laboratório, vocês
simplesmente tornam meus dias mais coloridos. Obrigadas especialmente àqueles que
contribuíram ativamente nas diferentes etapas deste trabalho.
Aos colegas do laboratório de biologia molecular do Serviço de Endocrinologia pelo
excelente ambiente de trabalho e pela cooperação em diversos momentos da execução desta
tese.
À querida amiga Patrícia Lopes pela paciência e pelo enorme auxílio teórico e prático
nos experimentos de citometria de fluxo.
À Profa. Dra Edna Teruko Kimura e ao Dr. César Seigi Fuziwara por terem
proporcionado um período de intenso aprendizado, pela paciência e pela efetiva contribuição
neste trabalho.
À minha amiga/irmã Alessandra Kherkoff pela amizade inestimável, pelas palavras
certas em todos os momentos e pelo apoio incondicional.
Aos meus pais, à minha irmã Francielle, ao meu sobrinho Lorenzo por todo o amor,
pelo carinho e pela força. Obrigada por vocês serem o alicerce da minha vida.
Ao meu companheiro Anderson Milani pelo amor, paciência, compreensão e apoio
incondicional.

3
Esta Tese de Doutorado segue o formato proposto pelo Programa de Pós-Graduação em
Ciências Médicas: Endocrinologia, Faculdade de Medicina, Universidade Federal do Rio
Grande do Sul, sendo apresentada na forma de manuscritos sobre o tema da Tese:
Artigo de revisão: Signaling Pathways in Follicular Cell-Derived Thyroid
Carcinomas (review); publicado no International Journal of Oncology. 2013 Jan;
42(1):19-28.
Artigo original: Type 3 deiodinase upregulation in papillary thyroid carcinoma is
mediated by crosstalk between MAPK and Sonic Hedgehog pathways and is
associated with cell proliferation
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
1
/
60
100%