Mémoire, DNF >

7
La Lettre du Neurologue - Suppl. Les Actualités au vol. IX - n° 9 - novembre 2005
ACTUALITÉS
neurosciences
neurosciences
>
ACTUALITÉS
neurosciences
neurosciences
> Behavioural Brain Research
> European Journal
of Neuroscience
> Nature
> NeuroImage
> Neuron
> Molecular psychiatry
>Science
> Trends in Neuroscience
> Neurobiology of Learning
and Memory
> PNAS
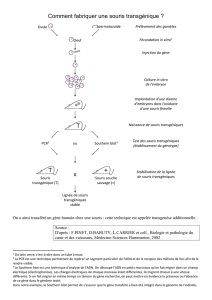
L
es lésions de dégénérescence
neurofibrillaire (DNF) sont prin-
cipalement composées de pro-
téines tau hyperphosphorylées. Leur
abondance est corrélée au déclin cogni-
tif et à la perte neuronale au cours de
la maladie d’Alzheimer et dans les
modèles murins de tauopathies. Cette
association entre DNF, perte neuronale
et troubles cognitifs chez l’homme et
l’animal a longtemps fait penser que les
lésions de DNF étaient à l’origine du
processus neurodégénératif et de la
dysfonction cérébrale. Toutefois, cette
hypothèse n’a jamais été testée de
façon rigoureuse. C’est désormais chose
faite grâce à une étude américaine au
cours de laquelle des souris transgé-
niques exprimant une forme mutée de
tau humain associée à des tauopathies
(P301L) est susceptible d’être réprimée
expérimentalement par la doxycycline.
Ces souris développent avec l’âge des
DNF, une perte neuronale ainsi que des
troubles du comportement et de la
mémoire. L’administration de doxycy-
cline, qui inhibe l’expression du trans-
gène tau, permet une amélioration des
performances cognitives des souris, une
stabilisation de leur perte neuronale
mais, de façon surprenante, les DNF
continuent de s’accumuler (1).
Commentaire
Cet article bouscule donc l’idée commu-
nément admise de l’existence d’un lien
direct entre déclin cognitif, mort cellu-
laire et lésions de DNF au cours des
Mémoire, DNF
et protéine tau :
un ménage à trois pas si simple…
tauopathies. Cela relance la théorie
suggérant que les DNF ne sont pas
forcément neurotoxiques. Deux revues
avancent des hypothèses quant aux
résultats “déstabilisants” de cet article.
Tout d’abord, dans l’éditorial qui
l’accompagne, V. Lee et J. Trojanowski
proposent que l’effet pathogène de
tau ne soit pas en rapport avec un gain
de fonction, mais avec une perte de
fonction qui perturbe le transport
axonal (2). Une autre synthèse récente
sur la phosphorylation et l’agrégation
anormale de tau envisage que les lésions
de DNF soient en fait neuroprotectrices
en réponse au stress oxydatif. Le débat
est donc ouvert sur les agrégats de pro-
téines tau dans les tauopathies : neuro-
protecteurs ou toxiques, responsables
d’un gain de fonction ou d’une perte de
fonction (3). La discussion s’étend aussi
aux autres pathologies neurodégénéra-
tives avec agrégats anormaux de pro-
téines, comme les synucléinopathies ou
la maladie de Huntington.
P. Derkinderen,
service de neurologie,
INSERM, Nantes.
Références bibliographiques
1.
Santa Cruz K et al. Tau suppression in a neurodege-
nerative mouse model improves memory function.
Science 2005;309:476-81.
2.
Trojanowski JQ, Lee VM. Pathological tau, a loss of
normal function or a gain in toxicity? Science 2005;
8:1136-7.
3.
Lee HG et al. Tau phosphorylation in Alzheimer’s
disease: pathogen or protector? Trends Mol Med 2005;
11:165-9.
Coordonné par P. Derkinderen
Service de neurologie, centre d’investigations cliniques
et INSERM U643, CHU de Nantes

ÉTRANGER (AUTRE QU’EUROPE)
FRANCE/DOM-TOM/EUROPE
Suppl. Vol. IX LN9
OUI, JE M’ABONNE AU MENSUEL La Lettre du Neurologue ET À SES SUPPLÉMENTS
ABONNEMENT : 1 an
+
➧➧➧➧➧➧➧ETPOUR 10 €DE PLUS !
❍10
€
accès illimité aux 24 revues de notre groupe de presse disponibles sur notre site
www.vivactis-media.com (adresse E-mail gratuite)
+
RELIURE
❍ 10
€
avec un abonnement ou un réabonnement
MODE DE PAIEMENT
❍
carte Visa, Eurocard Mastercard
N°
Signature : Date d’expiration
❍
chèque
(à établir à l’ordre de La Lettre du Neurologue)
❍
virement bancaire à réception de facture
(réservé aux collectivités)
Edimark SAS - 2, rue Sainte-Marie - 92418 Courbevoie Cedex
Rédacteur en chef
Pr Pierre Amarenco (Paris)
Total à régler .......... €
À remplir par le souscripteur
7ans
d’archivage
de nos revues
sur le site
Merci d’écrire nom et adresse en lettres majuscules
❍Collectivité ...........................................................................................................
à l’attention de .........................................................................................................
❍Particulier ou étudiant
M., Mme, Mlle ............................................................................................................
Prénom .......................................................................................................................
Pratique : ❍hospitalière ❍libérale ❍autre ..............................................
Adresse E-mail ..........................................................................................................
Adresse postale .......................................................................................................
..........................................................................................................................................
Code postal ................................Ville ……………………………………
Pays ..............................................................................................................................
Tél. .................................................................................................................................
Merci de joindre votre dernière étiquette-adresse en cas de réabonnement,
changement d’adresse ou demande de renseignements.
❍145
€collectivités
❍116
€particuliers
❍73
ێtudiants*
*joindre la photocopie de la carte
❍165
€collectivités
❍136
€particuliers
❍93
ێtudiants*
*joindre la photocopie de la carte

La Lettre du Neurologue - Suppl. Les Actualités au vol. IX - n° 9 - novembre 2005 9
D
ans la description initiale du syn-
drome de Gilles de la Tourette,
une origine familiale était suspectée…
Depuis, des hypothèses monogéniques
ou polygéniques ont été avancées, mais
rien n’a été prouvé. Un article fraîche-
ment publié dans Science devrait donc
faire parler de lui… Tout a commencé
par un patient de sexe masculin, dont
l’âge n’est pas précisé, qui présentait un
syndrome de Gilles de la Tourette, sans
antécédents familiaux, et dont l’analyse
du caryotype a montré une inversion sur
le chromosome 13. Une étude de géné-
tique moléculaire plus fine a permis de
montrer que 3 gènes candidats étaient
situés à proximité de la zone de “cassure”
à l’origine de l’inversion. Un seul de ces
gènes codait pour une protéine ayant
une fonction définie dans le système ner-
veux central, le gène Slit and Trk-like 1
(SLITRK1). La protéine SLITRK1 est, en
effet, abondante dans le système ner-
veux central et est impliquée dans la
croissance axonale. Les auteurs ont
ensuite screené 174 patients atteints de
syndrome de Gilles de la Tourette à la
recherche d’une mutation sur le gène
SLITRK1. Un patient présentait une muta-
tion qui décalait le cadre de lecture et
qui entraînait la production d’une forme
tronquée de la protéine. Cette même
mutation était aussi trouvée chez la
mère du patient, qui avait une tricho-
tillomanie. Cette mutation était absente
chez les 3 600 sujets témoins. Lorsque la
forme mutée de SLITRK1 est surexprimée
dans des neurones en cultures primaires,
elle ne favorise pas la croissance axonale,
contrairement à ce que l’on observe
avec la forme sauvage de la protéine.
Deux autres sujets testés, sans notion
L
a maladie d’Alzheimer est caracté-
risée, entre autres, par la présence de
dépôts extracellulaires de peptide -amy-
loïde. Grâce aux modèles animaux (souris
transgéniques), il est désormais admis que
cette accumulation de peptide -amyloïde
provoque des anomalies de la plasticité
synaptique bien avant la mort neuronale.
Il est suggéré que ces anomalies de la
plasticité synaptique mettent en jeu les
récepteurs du glutamate, et tout parti-
culièrement les récepteurs NMDA, mais
cela n’a jamais été prouvé… Une publi-
cation récente montre que l’exposition de
neurones de rat en culture primaire au
peptide -amyloïde diminue le nombre de
récepteurs NMDA à la surface cellulaire,
probablement par augmentation de leur
endocytose. Ces résultats ont ensuite été
confirmés in vivo, puisque la quantité de
récepteur NMDA est diminuée de moitié
dans les cerveaux de souris transgéniques
surexprimant une forme mutée de l’APP
(amyloid precursor protein), qui sont un
modèle murin de la maladie d’Alzheimer.
Un traitement de ces souris par un inhi-
biteur de la -sécrétase qui bloque la
production du peptide -amyloïde sup-
prime cet effet, montrant ainsi que la
baisse de la quantité de récepteurs NMDA
est bien la conséquence de la présence
du peptide -amyloïde.
Commentaire
Cet article permet donc de proposer un
mécanisme moléculaire précis et origi-
nal par lequel le peptide -amyloïde
serait toxique et néfaste pour la plasti-
cité synaptique et donc la mémoire à
un échelon cellulaire.
P. Derkinderen
>
Snyder EM et al. Regulation of NMDA receptor
trafficking by amyloid-. Nat Neurosci 2005;
8:1051-8.
de liens familiaux, avaient une particu-
larité dans la région non codante du
gène SLITRK1 qui aurait pour consé-
quence de diminuer l’expression de SLI-
TRK1.
Commentaire
Ce travail montre donc de façon convain-
cante qu’une mutation de SLITRK1 peut
être à l’origine d’un syndrome de Gilles
de la Tourette. Même si cette mutation
semble rare sur l’ensemble des syndromes
de Gilles de la Tourette, sa découverte
devrait permettre une meilleure compré-
hension de la physiopathologie de ce syn-
drome, comme cela a été le cas après la
découverte de mutations du gène codant
pour la synucléine dans la maladie de
Parkinson.
P. Derkinderen
>
Abelson et al. Sequence variants in SLITRK1
are associated with Tourette’s syndrome. Science
2005;310:317-20.
D
epuis le début des années 1960,
il a été montré que, chez le rat
adulte, des cellules souches situées
dans la zone sous-épendymaire du
ventricule latéral donnent perpétuel-
lement naissance à des précurseurs
neuronaux, qui migrent jusqu’au bulbe
olfactif (BO). On sait depuis qu’ils s’y
différencient en interneurones et que
leur intégration au réseau des récep-
teurs sensoriels est nécessaire à la
mémoire et à la discrimination olfac-
tive. Mais le dogme de l’immuabilité du
tissu nerveux chez l’adulte était si fort
que la communauté scientifique allait
Le peptide -amyloïde gêne
le trafic du récepteur NMDA
>
Enfin un gène
pour le syndrome
de Gilles de la Tourette ?
>
Les cellules souches
neurales adultes : un espoir
pour le traitement des maladies
neurodégénératives
>

La Lettre du Neurologue - Suppl. Les Actualités au vol. IX - n° 9 - novembre 2005
ACTUALITÉS
neurosciences
neurosciences
>
10
Coordonné par P. Derkinderen
> Behavioural Brain Research
> European Journal
of Neuroscience
> Nature
> NeuroImage
> Neuron
> Molecular psychiatry
>Science
> Trends in Neuroscience
> Neurobiology of Learning
and Memory
> PNAS
L
es phosphorylations sont les prin-
cipales modifications post-traduc-
tionnelles des protéines. La phosphory-
lation réversible de protéines est la
principale stratégie utilisée pour les
cellules eucaryotes afin de contrôler
l’activité des protéines. Dans les cel-
lules eucaryotes, trois acides aminés,
les résidus sérine, thréonine et tyro-
sine, peuvent être phosphorylés.
Plusieurs travaux ont montré que
l’alpha-synucléine était phosphorylée
sur sérine (en position 129) au sein des
corps de Lewy, mais le rôle de cette
phosphorylation dans le processus neu-
rodégénératif n’est pas connu. Une
équipe spécialisée dans les drosophiles
transgéniques aborde ce sujet dans un
récent numéro de Nature Neuroscience.
Dans un précédent travail, en surexpri-
mant de la synucléine humaine sauvage
ou mutée chez des drosophiles, la
même équipe avait pu obtenir un
modèle de la maladie de Parkinson chez
cette mouche avec la présence de diffi-
cultés motrices (difficultés à grimper le
long d’un tube à essai) et d’anomalies
histologiques (dégénérescence des
quelques neurones dopaminergiques
avec présence d’agrégats intraneuro-
naux). Afin d’étudier le rôle de la phos-
phorylation de l’alpha-synucléine au
cours du processus neurodégénératif,
ils ont réalisé des expériences similaires
avec des mutants de synucléine ne pou-
vant être phosphorylés et avec des
mutants de synucléine mimant une
phosphorylation constante (en rempla-
çant le résidu sérine par un résidu acide
aspartique). Les drosophiles surexpri-
mant la forme non phosphorylable de la
synucléine ne développent pas d’ano-
longtemps rejeter ou ignorer ces tra-
vaux fondateurs. Plus de vingt ans après,
la démonstration d’une neurogenèse
adulte chez l’oiseau chanteur, chez le
petit mammifère puis chez l’homme
allait susciter une formidable accélé-
ration des recherches. Le groupe de
Pierre-Marie Lledo avait déjà montré en
2004 le rôle clé de la ténascine-R dans
la migration des précurseurs neuronaux.
Un article récent de la même équipe
souligne le rôle déterminant du facteur
de transcription Pax6, qui contrôle
à la fois le niveau de neurogenèse
adulte et la différenciation des pro-
géniteurs. Pax6 engage d’abord les cel-
lules souches vers un destin neuronal.
Le maintien de son expression dans
les précurseurs neuronaux les oriente
ensuite vers un phénotype dopaminer-
gique, alors que son extinction induit
un phénotype GABAergique. La surex-
pression de Pax6 par un vecteur viral
provoque une augmentation considé-
rable du nombre de neurones dopami-
nergiques dans le BO.
Commentaire
Ces découvertes récentes sur les méca-
nismes de la migration et de la diffé-
renciation des cellules souches adultes
suscitent un formidable espoir pour le
traitement des maladies neurodégéné-
ratives comme la maladie de Parkinson.
Elles laissent entrevoir la possibilité de
détourner les cellules souches neurales de
leur destin olfactif vers d’autres structures,
telles que le striatum ou la substance
noire, et de les y différencier en neurones
dopaminergiques, ce qui permet d’éviter
les difficultés éthiques, techniques ou
immunologiques liées à l’utilisation de
cellules souches exogènes.
T. Lebouvier,
INSERM U643, Nantes.
>
Hack MA, Saghatelyan A, de Chevigny A et al.
Neuronal fate determinants of adult olfactory bulb
neurogenesis. Nat Neurosci 2005;8(7):865-72.
malies motrices ni de dégénérescence
des neurones dopaminergiques, alors
que celles surexprimant la forme “phos-
pho-mimétique” ont une forme accélé-
rée de maladie neurodégénérative. De
façon remarquable, les drosophiles qui
surexpriment la forme non phosphory-
lable de la synucléine ont plus d’inclu-
sions intraneuronales que les droso-
philes témoins et surexprimant la forme
“phospho-mimétique”.
Commentaire
Ces expériences montrent donc que la
phosphorylation sur sérine 129 de la
synucléine est essentielle au déve-
loppement du processus neurodégéné-
ratif. L’autre conclusion de ce travail est
que la présence d’inclusions intraneuro-
nales serait une réaction des neurones
au processus neurodégénératif ; ces
inclusions seraient neuroprotectrices
et non neurotoxiques, une idée très
“tendance” dans le domaine des patho-
logies neurodégénératives… À suivre.
P. Derkinderen
>
Chen L, Feany MB. Alpha-synuclein phospho-
rylation controls neurotoxicity and inclusion
formation in a Drosophila model of Parkinson
disease. Nat Neurosci 2005;8:657-63.
L
e glutamate est le principal neuro-
transmetteur excitateur du système
nerveux central. Il joue un rôle essentiel
en physiologie neuronale en étant, en
particulier, impliqué dans de nombreux
phénomènes de plasticité synaptique.
Toutefois, il est désormais admis que le
glutamate peut, dans certaines circons-
tances, être toxique pour les neurones,
Les bêta-lactamines
neuroprotectrices,
qui l’aurait prédit ?
>
La phosphorylation
de la synucléine est essentielle
pour sa neurotoxicité
>

La Lettre du Neurologue - Suppl. Les Actualités au vol. IX - n° 9 - novembre 2005 11
>
Rothstein JD, Patel S, Regan MR et al.
Beta-lactam antibiotics offer neuroprotection
by increasing glutamate transporter expression.
Nature 2005;433(7021):73-7.
L
es tauopathies, dont font partie la
maladie d’Alzheimer, la paralysie
supranucléaire progressive, la dégéné-
rescence cortico-basale et la maladie de
Pick, sont caractérisées par des lésions
intraneuronales appelées “dégénéres-
cence neurofibrillaire” (DNF). Le princi-
pal constituant des DNF est la protéine
tau présente sous forme d’agrégats,
d’où l’éponyme tauopathie. Au sein des
lésions de DNF, la protéine tau est anor-
malement phosphorylée et de nombreux
travaux suggèrent que cette phosphory-
lation joue un rôle dans l’agrégation de
tau. Une des principales enzymes capables
de phosphoryler tau est la glycogen syn-
thase kinase 3
(GSK3). L’utilisation
d’inhibiteurs de cette enzyme pourrait
donc s’avérer intéressante dans le trai-
tement des tauopathies.
Plusieurs récents travaux in vitro et in
vivo ont montré que le lithium, pour
des concentrations voisines de celles
utilisées dans le traitement des troubles
de l’humeur, est un inhibiteur de GSK3.
Afin d’étudier si l’inhibition de GSK3
pouvait être proposée comme traitement
potentiel des tauopathies, des souris
transgéniques surexprimant une forme
mutée de la protéine tau humaine ont
été traitées par lithium. Ces souris ont
déjà été caractérisées antérieurement
et présentent des déficits cognitifs
ainsi que des inclusions intraneuronales
constituées de protéine tau hyperphos-
phorylée. Les auteurs ont tout d’abord
vérifié que le traitement par lithium
inhibait bien la GSK3sérique chez les
comme cela est proposé dans la physio-
pathologie de la sclérose latérale amyo-
trophique. Une fois libéré dans l’espace
synaptique, le glutamate peut se fixer sur
ses récepteurs spécifiques ou être capté
par les astrocytes grâce à un transpor-
teur spécifique appelé GLT1. Des travaux
réalisés chez l’animal de laboratoire ont
montré que ce transporteur jouait un
rôle clé dans la régulation des fonctions
physiologiques et pathologiques du glu-
tamate. Aucun traitement n’était jusqu’à
présent connu pour moduler l’activité de
GLT1 ou modifier son niveau d’expres-
sion. En utilisant un modèle de tranches
de moelle épinière de rat, une équipe
de Baltimore a donc screené plus de
1000 molécules, à la recherche d’un
traitement capable de modifier l’expres-
sion de la protéine GLT1. Sur les
1040 traitements testés, les plus actifs
sur l’augmentation de l’expression de
GLT1 étaient les bêta-lactamines (péni-
cilline, ampicilline, amoxicilline, etc.).
Ces données ont été confirmées in vivo
chez la souris puisqu’un traitement
chronique par ceftriaxone augmente
l’expression de GLT1 dans l’hippocampe
et la moelle épinière.
Commentaire
Le dernier point de cet article, et vrai-
semblablement le plus remarquable,
est que la ceftriaxone a un effet chez
des souris transgéniques qui sont un
modèle murin de sclérose latérale
amyotrophique. La ceftriaxone adminis-
trée aux souris en début de maladie
permet non seulement de diminuer la
perte de poids et le déficit moteur, mais
aussi d’augmenter la durée de vie
moyenne des souris de 122 à 132 jours.
Un protocole de recherche multicen-
trique sur les effets de la ceftriaxone
chez des patients atteints de sclérose
latérale amyotrophique vient de débu-
ter (N Engl J Med 2005;352:13).
P. Derkinderen
souris. Ils ont ensuite montré, chez ces
souris transgéniques, que le lithium
diminuait la phosphorylation de tau sur
des épitopes habituellement phospho-
rylés par GSK3et que les quantités de
“tau agrégée” étaient moins importantes
chez les souris traitées.
Commentaire
Le lithium semble donc potentiellement
intéressant dans les tauopathies. Deux
petits bémols toutefois : l’effet sur les
troubles de la mémoire et sur les diffi-
cultés motrices des souris n’a pas été
étudié dans l’article, et l’on regrettera
qu’il n’y ait pas eu d’analyse plus appro-
fondie des sites de phosphorylation qui
sont sous la dépendance de cdk5.
P. Derkinderen
>
Noble W et al. Inhibition of glycogen syn-
thase kinase-3 by lithium correlates with reduced
tauopathy and degeneration in vivo. Proc Natl
Acad Sci USA 2005;102:6990-5.
Q
uatre types de muscles squelet-
tiques ont été étudiés : les muscles
deltoïde, quadriceps, gastrocnémien et
tibial antérieur. Trente-cinq échantillons
musculaires provenant de neuf patients
ont été prélevés lors d’autopsies. Les
hybridations des ARN correspondants
ont été réalisées sur des puces pangé-
nomiques Affymétrix (U133A, U133B).
Après une première étape de normalisa-
tion, trois facteurs apparaissent comme
discriminants dans la classification des
échantillons : l’âge, le sexe et le type de
muscle. Après une deuxième étape de
normalisation, seules les variations
observées en fonction du type de
muscle sont discriminantes. Les profils
De nouvelles perspectives
thérapeutiques pour le lithium ?
>
Les muscles squelettiques
humains possèdent des profils
d’expression de gènes différents
>
 6
6
1
/
6
100%