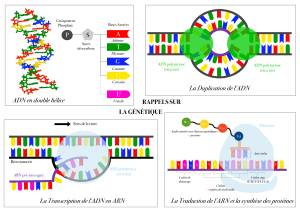
Amplification et séquençage

301
8. Amplification et séquençage des
acides nucléiques
ADN chromosomique, plasmides
(ADN bactérien), ARN messager
Rupture cellulaire
Isolation et purification
des acides nucléiques
Concentration et amplification et
séquençage de l’ADN ou de
l’ARN isolé

302
8.1 Extraction et isolation de l’ADN
(D’après genetics.nbii.gov/ basic2.html)

303
Extraction et isolation de l’ADN
Échantillon de cellules ou de tissus
Traitement avec une solution hypotonique
ou avec des détergents (Triton X-100 et SDS)
Rupture cellulaire et
dissociation des
complexes ADN-protéine
Incubation séquentiel avec un enzyme
protéolytique (protéinase K) et la ribonucléase Élimination de protéines
et d’ARN
Extraction de l’ADN dans de l’éthanol
Seules les longues chaînes
d’ADN précipitent dans l’EtOH;
les nucléotides simples et les
produits de la digestion de l’ARN
restent dans la phase aqueuse.

304
Centrifugation par gradient de densité:
Une méthode alternative pour l’isolation de
l’ADN est la séparation de protéines, ARN
et ADN selon des différences dans leurs
densités flottantes ou poussées.
Une telle séparation est effectuée en
centrifugeant à haute vitesse l’échantillon
brut dans une éprouvette contenant un
gradient de densité formé (ex. 1.6 à 1.8
g/mL) par une solution de CsCl.
Lors de la centrifugation, les
macromolécules (protéines, ADN, ARN)
présentent dans la solution de CsCl forment
des bandes distinctes selon leurs densités
flottantes. (D’après D. Holme et H. Peck,
Analytical Biochemistry, 3è éd., 1998)

305
L’ADN chromosomique peut être séparé des plasmides par centrifugation par
gradient de densité en présence du bromure d’éthidium (EtBr).
L’EtBr est un fluorophore qui se lie entre deux brins d’ADN. En se liant à
l’ADN, l’EtBr diminue la densité flottante de celle-ci, dû à un déroulement
partiel de la double hélice.
Il y a plus d’EtBr qui se lie à l’ADN chromosomique (ADN linéaire) qu’aux
plasmides (ADN circulaire).
La densité de l’ADN chromosomique est diminuée par 0.125 g/mL, tandis que
que celle des plasmides est diminuée par 0.085 g/mL.
(D’après D. Holme et H. Peck,
Analytical Biochemistry, 3è éd., 1998)
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
1
/
44
100%