Lire l'article complet

Médecine
& enfance
CAS CLINIQUE
Morgane, accompagnée de sa maman,
se présente à la consultation de diabéto-
logie pédiatrique pour exploration
d’une hyperglycémie postprandiale au-
tour de 1,5 g/l. Elle est née à terme, eu-
trophique, et n’a pas d’antécédents per-
sonnels particuliers. Sa maman est sui-
vie pour un diabète de type MODY 2,
confirmé par l’analyse génétique (muta-
tion constitutionnelle sur la séquence
codante du gène GCK). Elle n’a pas de
traitement particulier et respecte une
alimentation équilibrée. Le grand-père
de Morgane a également un diabète
MODY 2, confirmé, traité par antidiabé-
tiques oraux. L’oncle maternel a aussi
un diabète MODY 2 non traité.
Morgane est en très bon état général. Le
bilan sanguin effectué trouve une hé-
moglobine glyquée à 6,8 % (N : 4-6 %).
Les anticorps anti-GAD, anti-îlots de
Langerhans et anti-IA2 sont négatifs. La
prise en charge du diabète de Morgane
passe par des conseils diététiques
simples : manger équilibré et éviter les
excès de glucides. La confirmation du
diabète MODY 2 est obtenue par la mise
en évidence de la délétion du gène de la
glucokinase identifiée chez la maman et
le grand-père maternel. L’hémoglobine
glyquée de Morgane restera égale à
6,5 % avec de simples mesures diété-
tiques. Sa croissance est satisfaisante et
elle n’a pas d’hyperglycémies matinales
ou postprandiales.
Cette présentation clinique est fréquen-
te en France, en raison de la prédomi-
nance du MODY 2 ; le diagnostic est
souvent fortuit ou posé lors d’un bilan
génétique dans une famille connue por-
teuse de la mutation.
CRITÈRES DIAGNOSTIQUES
DU DIABÈTE MODY
Le diabète MODY est défini comme
étant une forme familiale juvénile de
diabète de type 2 qui apparaît dans l’en-
fance, l’adolescence ou chez l’adulte
jeune [4].
Le diagnostic se fonde sur cinq critères
majeurs :
첸
hyperglycémie diagnostiquée avant
l’âge de vingt-cinq ans chez un ou deux
membres d’une même famille ;
첸
hérédité : transmission autosomique
dominante sur trois générations ;
FMC DE NÎMES
Les diabètes monogéniques:
les formes MODY
R. Salet, service de pédiatrie,
CHU Carémeau, Nîmes
mars 2013
page 69
Rubrique dirigée par T.A. Tran, service
de pédiatrie, CHU Carémeau, Nîmes
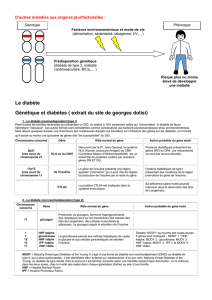
Le diabète se traduit par l’existence d’une hyperglycémie chronique. Les deux
formes les plus fréquentes sont le diabète de type 2 (80 %), dont l’étiologie
est multifactorielle, et le diabète de type 1 (15 %), d’origine auto-immune par
destruction des cellules pancréatiques sécrétant l’insuline. Le diabète MODY
(maturity onset diabetes of the young), qui représente 2 à 5 % des diabètes [1],
est une forme monogénique familiale du diabète de type 2, de transmission
autosomique dominante et à début précoce, survenant chez l’enfant, l’adoles-
cent ou l’adulte jeune. Au cours des années 1990, six gènes associés à ce ty-
pe de diabète ont été identifiés [2, 3]. Ils sont tous impliqués dans la cascade
d’événements aboutissant à la sécrétion d’insuline. Les formes les plus fré-
quentes sont les MODY 2 et 3. Le diagnostic peut rester longtemps méconnu
en raison de l’hyperglycémie modérée et non symptomatique, mais les compli-
cations à long terme sont identiques à celles observées dans le diabète auto-
immun de type 1, d’où la nécessité de reconnaître ce type de diabète pour per-
mettre sa prise en charge précoce.
69-71_xpr8 21/03/13 00:05 Page69

첸
pas de traitement par insuline cinq
ans après le diagnostic ou taux de pepti-
de C important chez un patient sous in-
suline ;
첸
insulinémie normale ou anormale-
ment basse par rapport à l’hyperglycé-
mie ;
첸
absence de surpoids ou d’obésité.
LES DIFFÉRENTS TYPES
DE MODY ET LEUR PRISE
EN CHARGE
MODY 1
Ce type de diabète est dû à des muta-
tions hétérozygotes du gène HNF4aet
représente 20 % des diabètes MODY en
France. Il apparaît entre sept et quarante
ans. L’hyperglycémie à jeun est présente
chez 95 % des sujets porteurs de la mu-
tation. Cliniquement, ce diabète ne se
distingue pas du MODY 3. La mutation
entraîne un trouble de la sécrétion d’in-
suline par les cellules bde Langerhans
ainsi que du transport intracellulaire du
glucose et de son métabolisme [5, 6].
La mutation HNF4aserait associée à
une macrosomie néonatale et à un
poids de naissance élevé, ce qui établit
le rôle du HNF4adans la régulation in
utero de la sécrétion pancréatique d’in-
suline et dans la détermination du poids
de naissance [7].
MODY 2
Le diabète MODY 2 représente 50 % des
diabètes monogéniques ; c’est la forme
la plus fréquente en France. Il est dû à
une mutation à l’état hétérozygote du
gène de la glucokinase, enzyme respon-
sable de la transformation dans les cel-
lules bde Langerhans et dans le foie du
glucose en glucose-6-phosphate. Ce mé-
canisme régule la sécrétion de l’insuli-
ne. Plus de cent cinquante mutations
ont été décrites à ce jour.
Dans ce type de diabète, le seuil de gly-
cémie qui déclenche la sécrétion d’insu-
line est élevé, d’où l’hyperglycémie mo-
dérée mais permanente [8].
Le diagnostic repose sur l’histoire fami-
liale. La pénétrance de la mutation est
complète : tous les porteurs de la muta-
tion ont une hyperglycémie.
Dans le cas où ce diabète est méconnu
dans une famille, le diagnostic est sou-
vent posé de façon fortuite lors d’un bi-
lan sanguin, en l’absence de tout signe
clinique. Certaines situations de stress
(chirurgie, fièvre, maladies infectieuses
ou corticothérapie) peuvent révéler l’hy-
perglycémie et conduire au diagnostic.
MODY 3
Moins fréquent que le MODY 2 en Fran-
ce, où il représente 20 % des diabètes
génétiques du jeune, son incidence se-
rait de 60 % au Royaume-Uni et de
50 % aux Etats-Unis. Il est dû à une mu-
tation à l’état hétérozygote du gène
HNF1a, dont l’expression phénotypique
peut être différente d’un individu à
l’autre : dans une même famille, cer-
tains sujets porteurs de la mutation
peuvent être normoglycémiques alors
que leurs frères et sœurs ou cousins
peuvent être hyperglycémiques [9]. Le
diagnostic se fait généralement à la pé-
riode postpubertaire ; il est souvent pré-
cédé d’un syndrome polyuro-polydip-
sique avec perte de poids.
MODY 4
Très rare en France, le MODY 4 est dû à
Médecine
& enfance
mars 2013
page 70
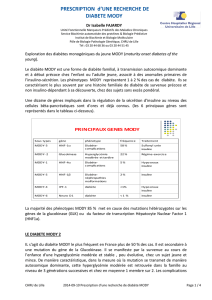
Les gènes du diabète MODY
Type de MODY Anomalies génétiques Localisation Année
Anomalies génétiques chromosomique de découverte
MODY 1 Mutations du gène HNF4aChromosome 20 1991
MODY 2 Mutation du gène de la glucokinase Chromosome 7 1992
MODY 3 Mutation du gène HNF1aChromosome 12 1994
MODY 4 Mutation du gène IPF1 Chromosome 13 1997
MODY 5 Mutation du gène HNF1bChromosome 17 1997
MODY 6 Mutation NeuroD1 Chromosome 2 1999
HNF : hepatocyte nuclear factor
IPF : insulin promoter factor
NeuroD1 : neurogenic differenciation factor 1
Manifestations cliniques et traitement
Type Fréquence Premières Phénotype Clinique Traitement
de MODY conséquences clinique et premières
anomalies
MODY 1 Pancréas Hyperinsulinisme Diabète Traitement
néonatal Complications hypoglycémiant
micro- et macro-
vasculaires
MODY 2 10 à 60 % Pancréas Hyperglycémie Prévalence faible Alimentation
France et foie modérée des complications équilibrée
et Italie et tardive microvasculaires
MODY 3 20 à 65 % Pancréas Diabète Complications Traitement
Angleterre et rein micro- et macro- hypoglycémiant
vasculaires Sulfamide
hypoglycémiant
Insuline
MODY 4 Pancréas Diabète
MODY 5 Rein Diabète Hypoplasie Insuline
et pancréas pancréas
69-71_xpr8 21/03/13 00:06 Page70

une mutation du gène IPF1 à l’état hété-
rozygote. Ce gène associé à celui de l’in-
suline a un rôle fondamental dans le dé-
veloppement des cellules βdu pan-
créas. En cas de mutation à l’état homo-
zygote, une agénésie pancréatique res-
ponsable de diabète néonatal et d’une
insuffisance pancréatique exocrine est
possible [10-11]. Le MODY 4 se révèle
plus tardivement que les formes habi-
tuelles de MODY.
MODY 5
Le MODY 5, dû à des mutations du gène
HNF1β, est associé à des anomalies ré-
nales constantes, morphologiques et
fonctionnelles (kystes, insuffisance ré-
nale chronique progressive), ainsi qu’à
certaines anomalies du développement
génital [12].
MODY 6
Très rare, il est dû à une mutation Neu-
roD1, le facteur primordial dans le dé-
veloppement du pancréas et du système
nerveux central. Il régule l’expression
du gène de l’insuline, et son déficit en-
traîne une anomalie de sécrétion [13].
QUAND FAUT-IL ÉVOQUER
LE DIABÈTE MODY ?
Le diagnostic génétique du MODY n’est
pas de pratique courante. Il s’agit d’une
analyse coûteuse et fastidieuse. Le dia-
gnostic doit être évoqué dans des situa-
tions spécifiques : diabète survenant chez
des sujets ayant une histoire familiale
concernant trois générations, de révéla-
tion précoce, en l’absence de surpoids.
L’histoire clinique oriente ensuite vers le
gène à étudier : MODY 2 en cas d’hyper-
glycémie modérée, MODY 3 en cas de
présentation sous forme d’acidocétose,
MODY 5 en cas d’anomalies rénales.
L’identification de ces gènes permet une
amélioration de la prise en charge des
patients, fondée sur la connaissance du
pronostic, des complications au long
cours et du traitement adapté.
첸
Références
[1] FAJANS S.S., BELL G.I., POLONSKY K.S. : « Molecular mecha-
nisms and clinical pathophysiology of maturity-onset diabetes of
the young »,
N. Engl. J. Med.,
2001 ;
345 :
971-80.
[2] PEARSON E.R., VELHO G., CLARK P. et al. : « ß-cell genes
and diabetes : quantitative and qualitative differences in the pa-
thophysiology of hepatic nuclear factor-1αand glucokinase mu-
tations »,
Diabetes,
2001 ;
50 :
S101-7.
[3] VELHO G., FROGUEL P. : « Maturity-onset diabetes of the
young (MODY), MODY genes, and non-insulin-dependent dia-
betes mellitus »,
Diabetes Metab.,
1997 ;
23 (suppl. 2) :
34-7.
[4] VAXILLAIRE M., FROGUEL P. : « Monogenic diabetes in the
young, pharmacogenetics and relevance to multifactorial forms
of type 2 diabetes »,
Endocr. Rev.,
2008 ;
29 :
254-64.
[5] STOFFEL M., DUNCAN S.A. : « The maturity-onset diabetes
of the young (MODY) transcription factor HNF4αregulates ex-
pression of genes required for glucose transport and metabo-
lism »,
Proc. Natl Acad. Sci. USA,
1997 ;
94 :
13209-14.
[6] GUPTA R.K., VATAMANIUK M.Z., LEE C.S. et al. : « The MO-
DY1 gene HNF-4αregulates selected genes involved in insulin
secretion »,
J. Clin. Invest.,
2005 ;
115 :
1006-15.
[7] PEARSON E.R., BOJ S.F., STEELE A.M. et al. : « Macrosomia
and hyperinsulinaemic hypoglycemia in patients with heterozy-
gous mutations in the HNF4αgene »,
PLoS Med.,
2007 ;
4:
e118.
[8] VELHO G., PETERSON K.F., PERSEGHIN G. et al. : « Impaired
hepatic glycogen synthesis in glucokinase-deficient (MODY-2)
subjects »,
J. Clin. Invest.,
1996 ;
98 :
1755-61.
[9] VAXILLAIRE M., PUEYO M.E., CLEMENT K. et al. : « Insulin se-
cretion and insulin sensitivity in diabetic subjects with hepatic
nuclear factor-1α(maturity-onset diabetes of the young 3) muta-
tions »,
Eur. J. Endocrinol.,
1999 ;
141 :
609-18.
[10] STOFFERS D.A., FERRER J., CLARKE W.L. et al. : « Early-on-
set type-II diabetes mellitus (MODY4) linked to IPF1 »,
Nat. Ge-
net.,
1997 ;
17 :
138-9.
[11] MAESTRO M.A., CARDALDA C., BOJ S.F. et al. : « Distinct
roles of HNF1β, HNF1α, and HNF4αin regulating pancreas de-
velopment, β-cell function and growth»,
Endocr. Dev.,
2007 ;
12 :
33-45.
[12] HAUMAITRE C., BARBACCI E., JENNY M. et al. : « Lack of
TCF2/vHNF1 in mice leads to pancreas agenesis »,
Proc. Natl
Acad. Sci. USA,
2005 ;
102 :
1490-5.
[13] LIU L., FURUTA H., MINAMI A. et al. : « A novel mutation,
Ser159Pro, in the NeuroD1/β2 gene contributes to the develop-
ment of diabetes in a Chinese potential MODY family »,
Mol.
Cell. Biochem.,
2007 ;
303 :
115-20.
Médecine
& enfance
mars 2013
page 71
05 ma13 m&e nîmes diabète 21/03/13 11:46 Page71
1
/
3
100%