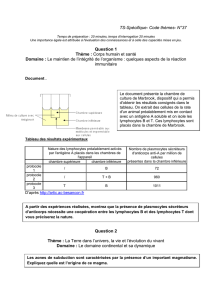
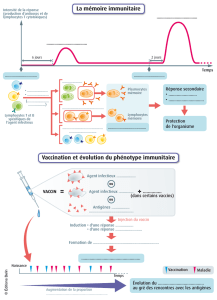
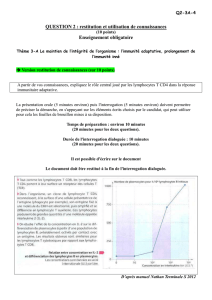
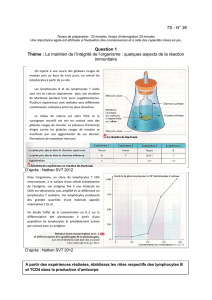
Étude de la différenciation in vitro de lymphocytes

I
Étude sur la différenciation in vitro de lymphocytes B
humains en plasmocytes
Microenvironnement et caractérisation des plasmocytes générés
Mémoire
Rayelle Itoua Maïga
Maîtrise en biochimie
Maître ès sciences (M.Sc.)
Québec, Canada
© Rayelle Itoua Maïga, 2013


III
Résumé
Les plasmocytes, cellules responsables de la sécrétion d’anticorps, sont au cœur de la réaction immunitaire humorale.
Cette caractéristique en fait des cellules intéressantes en thérapie cellulaire pour offrir une protection humorale aux
patients immunosupprimés suite à une transplantation de cellules souches hématopoïétiques. Afin d’obtenir ces cellules,
notre équipe à Héma-Québec propose d’utiliser la capacité de différenciation des lymphocytes B mémoires in vitro. Notre
hypothèse est qu’il serait possible de différencier ces derniers en contrôlant leur microenvironnement de culture. Ce
projet porte donc sur l’étude de l’interaction cellulaire CD27-CD70 combinée aux facteurs de survie APRIL et CXCL12.
Les résultats montrent que cette interaction induit une différenciation rapide des lymphocytes B tandis que l’addition des
facteurs solubles n’a pas d’impact sur la survie cellulaire. De plus, l’utilisation des marqueurs de surface CD31 et CD39 a
permis de mettre en évidence l’hétérogénéité des plasmocytes générés in vitro et ceux retrouvés in vivo.


V
Table des matières
Résumé ........................................................................................................................ III
Liste des figures ........................................................................................................... IX
Liste des abréviations ................................................................................................... XI
Remerciements .......................................................................................................... XIII
1. Introduction ............................................................................................................. 1
1.1 Les lymphocytes B humains ............................................................................................... 1
1.1.1 Développement et transition .................................................................................................... 1
1.1.2 Maturation des lymphocytes B et rencontre de l’antigène ............................................... 1
1.1.3 Activation extrafolliculaire des lymphocytes B .................................................................. 2
1.1.4 Activation folliculaire et formation du centre germinatif .................................................. 2
1.1.5 Formation du centre germinatif ......................................................................................... 3
1.1.6 Différenciation terminale des lymphocytes B .................................................................... 5
1.1.7 Réponse immunitaire secondaire ...................................................................................... 6
1.1.8 Les populations de lymphocyte B du sang ......................................................................... 6
1.2 Les plasmocytes ............................................................................................................... 7
1.2.1 Les immunoglobulines ....................................................................................................... 7
1.2.2 Le phénotype des lymphocytes B différenciés .................................................................. 8
1.2.3 Plasmocytes à courte vie .................................................................................................. 10
1.2.4 Les plasmocytes à longue vie ........................................................................................... 10
1.2.5 Niche de survie des plasmocytes ..................................................................................... 11
1.3 Les plasmocytes et la thérapie cellulaire ......................................................................... 13
1.3.1 Greffe de cellules souches hématopoïétiques ................................................................. 13
1.3.2 Les applications de la transplantation ............................................................................. 14
1.3.3 La reprise de greffe et la reconstitution du système immunitaire .................................. 14
1.3.4 Problèmes post-transplantation et vulnérabilité aux infections ..................................... 15
1.3.5 Traitement des problèmes post-transplantations ........................................................... 15
1.3.6 Introduction des plasmocytes générés in vitro ................................................................ 16
1.4 Modèles de culture développés jusqu’à présent ............................................................. 17
1.5 Expansion et différenciation in vitro de lymphocytes B humains ..................................... 17
1.5.1 Interaction CD40-CD154 .................................................................................................. 17
1.5.2 Interaction CD27-CD70 .................................................................................................... 18
2. Hypothèse ................................................................................................................ 19
3. Objectifs .................................................................................................................. 21
4 Méthodologie ........................................................................................................... 23
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
1
/
96
100%