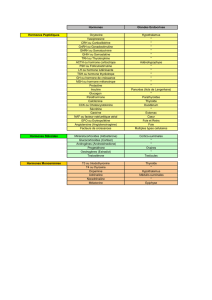
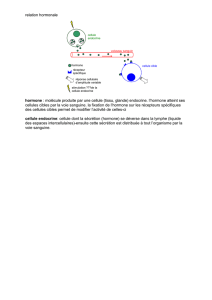
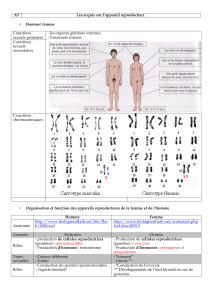
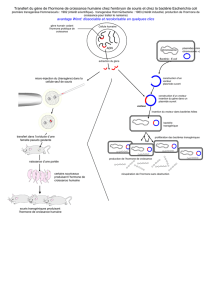
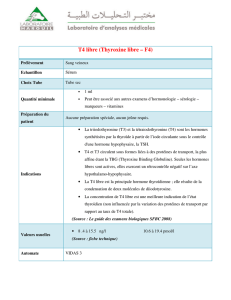
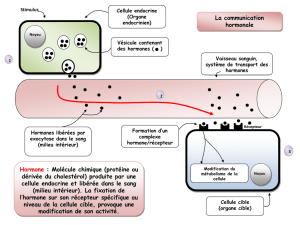
11- Approche génétique et biologique du retArd de croissance.

RÉPUBLIQUE ALGÉRIENNE DÉMOCRATIQUE ET POPULAIRE
MINISTÈRE DE L’ENSEIGNEMENT SUPÉRIEUR
ET DE LA RECHERCHE SCIENTIFIQUE
Université des Frères Mentouri Constantine
Faculté des Sciences de la Nature et de la Vie
Département de Biologie Animale
Mémoire présenté en vue de l’obtention du Diplôme de Master
Domaine : Sciences de la Nature et de la Vie
Filière : Sciences Biologiques
Spécialité : Génétique Moléculaire
Intitulé :
Jury d’évaluation :
Président du jury : Pr SATTA D (Professeur de l’université de Mentouri Constantine).
Rapporteur : Pr BENMEBAREK (Professeur de la faculté Médecine, Constantine).
Examinateurs : Pr BENMOUHAMED (Professeur de la faculté Médecine, Constantine).
Année universitaire
2014 – 2015
Approche génétique et biologique
du retArd de croissAnce
Présen
té
et soutenu par
:
Atrous Khaoula
Le
:
01
/0
7
/2015
Barkat Amina

Au Nom de Dieu, le Tout Miséricordieux, le
Très
Miséricordieux
Merci dieu tout puissant, qui m’a honoré d’être
parmi ceux qui savent lire et écrire, et qui a
guidé mes pas sur le chemin de la science.
Je l’implore de m’éclairer et de me guider sur le
droit chemin

Remerciement
C’est avec sincérité que nous remercions notre encadreur Pr
BENMEBAREK KARIMA qui a accepté avec toute modestie de nous encadrer,
pour sa disponibilité, ses conseils avisés, ses exigences, et son esprit critique qui
ont concouru à la réalisation de ce travail malgré ses multiples charges, tout le
long de l’année
Nous tenons à remercier la responsable de spécialité Mme SATTA pour sa
patience et surtout pour sa confiance, ses remarques et conseils, sa disponibilité
et sa bien vaillance.
On voudra également remercier les membres du jury pour avoir accepté
d’évaluer ce travail.
Ainsi que le personnel et les enseignements de la spécialité de génétique.
Nous tenons aussi à remercier toutes les techniciennes du laboratoire
d’hormonologie du CHU et nous remercions Dr BOUKHELKHAL Unité de
Cytogénétique et Dr KHENSAL Maitre - Assistante au service.
Nos remerciements touchent à la fin et c’est le moment de remercier tous les
membres de nos familles, en commençant par nos parents qui nous ont toujours
soutenus et dont la patience et l’encouragement nous ont été très précieux durant
nos années d’études.

Dédicaces
Ce travail modeste est dédié :
A mes parents,
Halima ;et Badaoui
A mes belles sœurs Chaima, Izdihar et
Nedjma
A tous mes familles
A tous ceux qui, par un mot, m’ont donné la
force de continuer …..
khaoula

Dédicaces
Ce travail modeste est dédié :
A mon père Hocine
À ma belle-mère Habiba
À la mémoire de mon grand-père El Bachir miséricorde de Dieu
À mes proches de mes frères Mourad, Achraf, Wassim et la fiancée de mon frère
Zeineb Boutteba
À toute la famille ; mes tentes et leurs maris
A mon oncle etSa famme
À tous mes amis ;
À tous mes chers enseignants qui ont enseigné moi ;
Amina
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
1
/
68
100%