UE 2V311 - TD 2

7
UE 2V311 - TD 2 - 2015
Structure des gènes.
Buts du TD :
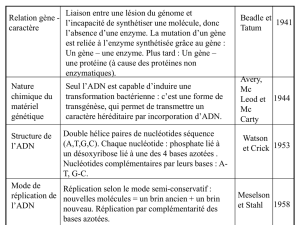
1) connaître les éléments de structure d’un gène eucaryote et procaryote (Rappel 1V002).
2) connaître les différences étapes de l’expression d’un gène (Rappel 1V002).
3) comprendre comment identifier des gènes par analyse informatique.
4) connaître les différents types de mutations et leurs conséquences.
Documents utiles : structure des gènes procaryotes et eucaryotes du poly de cours.
I. Travail personnel pour tester vos connaissances (non corrigé en TD) : Comparaison des
éléments de structure des gènes procaryotes et eucaryotes codant une protéine.
Remplissez le tableau ci-dessous en indiquant l’existence ou l’absence de l’élément (précisez sa
nature s’il existe) ainsi que le processus dans lequel il est impliqué. En prenant l’exemple d’un
gène eucaryote codant une protéine, positionnez sur un schéma ces différents éléments et
représentez les étapes de l’expression de ce gène.
Elément de structure
Gène procaryote
Gène eucaryote
Processus
Promoteur
Séquences régulatrices
+1 de transcription
Introns / Exons
Régions 5’UTR et
3’UTR
Terminateur de
transcription
Phase de lecture
ouverte (ORF / CDS)
Séquence RBS
Codon d’initiation
Codon stop

8
II. Décryptage de séquences nucléotidiques chez les eucaryotes.
Une façon d’identifier les gènes présents dans un génome à partir de la séquence de
celui-ci est de rechercher des éléments de structure d’un gène.
Q1. Est-il possible d’identifier, d’après leur séquence, tous les éléments cités dans le tableau
précédent ?
Q2. Recherchez dans la séquence double brin de 199 paires de bases (pb) ci-dessous certains
des éléments du tableau que vous aurez choisis. Par convention, le brin 5’→3’ est celui du
dessus.
Pour étudier des génomes de plusieurs millions (voire milliards) de paire de bases, on
utilise des logiciels tels que Ape (gratuit sur internet) pour effectuer des recherches
systématiques. Une des fonctions de ce logiciel est la recherche de phases ouvertes de lecture
(ORF, Open Reading Frame). Cette fonction appelée ORFMAP consiste à lire la séquence
d’ADN fournie à la manière d’un ribosome. En prenant les nucléotides 3 par 3, elle va
transformer la séquence en une image où chaque codon 5’-ATG-3’ trouvé sera indiqué par un
petit trait et chaque codon 5’-TAA-3’, 5’-TGA-3’, et 5’-TAG-3’ trouvé sera indiqué par un grand
trait. Les autres codons ne sont représentés par rien.
Q3. Sur une molécule d’ADN double brin, combien y a-t-il de phases de lecture possibles ?
La figure suivante montre le résultat de cette fonction ORFMAP sur une séquence de
6100 pb d’un organisme procaryote.
Prenons les règles d’analyse suivantes :
1) une ORF correspond à une phase ouverte de lecture comprise entre deux codons stop
2) une CDS (CoDing Sequence) correspond à une phase ouverte de lecture comprise
entre un ATG et un codon stop
3) une CDS ne sera retenue comme gène potentiel que si elle n’est pas incluse dans une
plus grande CDS et fait au moins 300 pb de longueur.

9
Q4. En respectant ces règles, identifiez les CDS correspondants à des gènes potentiels dans
la séquence présentée ci-dessus.
Q5. Faites un schéma représentant le fragment d'ADN étudié sous forme double brin
orientés et positionnez les éléments du tableau précédent, en précisant, si cela est
nécessaire, à partir de quel brin ils sont transcrits.
Q6. En théorie, quelle serait la longueur en acides aminés des protéines synthétisées à partir
de ces CDS ainsi que leur masse moléculaire (on rappelle que la masse moléculaire
moyenne d'un acide aminé est 110 daltons) ?
Q7. Comment vérifier expérimentalement que ces séquences correspondent bien à des
gènes ?
III. Relation entre la carte des ORFs et celle des gènes.
Q8. Pensez-vous que l’on puisse trouver tous les gènes d’un organisme grâce à cette fonction
ORFMAP si l’on connaît toute la séquence du génome de cet organisme ? Si non, quels sont
les types de gènes qui échappent à cette analyse ?
Q9. Comment faire pour identifier les gènes qui échappent à l’analyse par ORFMAP ?
Le gène TORP1 est impliqué dans différentes pathologies cardiaques et a été localisé sur
le bras court du chromosome 3 chez l’homme. La figure ci-dessous représente le profil
ORFMAP réalisée sur le fragment d’ADN génomique contenant le gène (A) ou celui réalisé sur
l’ADN complémentaire (ADNc) correspondant (B).
NB : les deux cartes ne sont pas exactement à la même échelle
Q10. Rappelez ce qu’est un ADNc et comment on l’obtient.
Q11. D’après le profil B, indiquez quel brin d’ADN a la même séquence que l’ARN
messager.
Q12. Expliquez par un schéma la différence entre la carte des ORFs obtenue sur le fragment
d’ADN génomique et celle obtenue à partir de l’ADNc.
>3
>2
>1
<1
<2
<3
A
B

10
IV. Les mutations et leurs conséquences.
A partir d'individus atteints de pathologies cardiaques, différentes mutations du gène
TORP1 ont été caractérisées et mises en relation directe avec le développement de la maladie.
Les coordonnées des mutations sont données par rapport au premier nucléotide du codon
d’initiation de la traduction
Q13. Complétez le tableau ci-dessous en vous aidant du code génétique fourni en annexe.
Patient
Mutation
sain→malade
Type
moléculaire de
mutation
Effet de la mutation
Conséquence sur la
protéine
GB001
Δ214-216AAG
GB003
274CAG→274TAG
RU001
457GAG→457GGG
RU018
760TAT→760TAA
JP084
Δ4A
Q14. Que donnerait le profil ORFMAP correspondant à l’ADNc de ces mutants ?
ANNEXE – Code génétique

11
FIN DU TD
Pour vous entrainer :
SUP1 : Analysez la séquence génomique dont la carte des ORF vous est montrée ci-dessous,
issue d'un organisme procaryote, en répondant aux questions Q4 à Q6.
SUP2 : Etes vous capable de dire rapidement à quoi correspond chaque numéro ?
Pour aller plus loin...
SUP3 : Sudoku génétique. Compléter le tableau suivant. Préciser quel est le brin codant de
l'ADN.
 6
6
1
/
6
100%