les accidents de decompression: physiopathologie

LES ACCIDENTS DE DECOMPRESSION:
PHYSIOPATHOLOGIE, TRAITEMENT
MEDICAMENTEUX.
Physiopathologie des accidents de décompression
J.-L. MELIET
Ecole d’application du service de santé des armées
1, place Alphonse Laveran, 75230 Paris Cedex 5
Tél. 01 40 51 43 72. Fax 01 40 51 47 38. e-mail : [email protected]
Traitement médicamenteux de l'accident et de la maladie de décompression.
I. ROUQUETTE- VINCENTI
Service d'Anesthésie- Réanimation
H.I.A. Val de Grâce, 34 Bd Port Royal, 75005 PARIS.

Plan.
I- PHYSIOPATHOLOGIE DE L'ACCIDENT DE DECOMPRESSION.
I- 1- L'accident bullaire initial.
I- 2- La maladie de décompression.
Interaction bulles paroi: Au niveau de la membrane basale.
Activation des cellules endothéliales.
Interaction bulles plasma.
Interaction avec les éléments figurés du sang.
Les plaquettes.
Les leucocytes.
Les hématies.
Mise en jeu du système immmunitaire.
I- 3- Les conséquences microcirculatoires.
Au niveau pulmonaire.
Au niveau systémique.
II- LE TRAITEMENT SYMPTOMATIQUE.
II- 1- Position.
II- 2- Oxygénothérapie.
II- 3- Remplissage vasculaire.
Réhydratation IV.
Réhydratation orale.
II- 4- Lutte contre l'hypothermie.
III- LE TRAITEMENT PHYSIOPATHOLOGIQUE.
III- 1- Glucocorticoïdes et A.I.N.S.
Glucocorticoïdes.
A.I.N.S.
Voie du complément.
III- 2- Antiagrégants et anticoagulants.
III- 3- Lidocaïne.
III- 4- Vasodilatateur.
III- 5- Agents tensioactifs.

III- 6- Anti ischémiques.
IV- CONCLUSION.

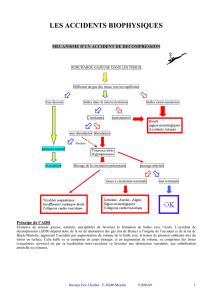
On sait depuis Paul Bert (1878) que les manifestations pathologiques qui surviennent à l'issue d'un
séjour en pression sont dues à la présence de bulles qui, embolisant le réseau vasculaire, sont à l’origine
d’ischémies le plus souvent neurologiques. En 1961, Laborit et Barthélémy décrivaient les manifestations
biologiques accompagnant ces accidents (amas plaquettaires, thromboses, vaso-constriction, stase
circulatoire, extravasation, œdèmes interstitiels), qui constituent la maladie de la décompression (1).
L’accident de décompression (ADD)comporte deux versants : l’accident bullaire initial et la maladie de la
décompression (MDD).
I- 1. L’accident bullaire initial
Sous pression, l'azote de l'air se dissout dans les liquides de l'organisme. Non métabolisé, il doit être
restitué à l'atmosphère lors du retour à la pression normale. Cette désaturation de l’organisme n'est pas
instantanée. Elle suit une loi exponentielle (Haldane, 1908) telle que la désaturation complète de
l’organisme n’est atteinte qu’en 12 heures environ. Des tables de plongée, maintenant calculées en continu
par des ordinateurs portables, donnent en fonction du temps passé aux différentes profondeurs la durée de
remontée et les temps d'arrêt à respecter à chaque palier. Le non-respect de cette cinétique expose au
développement de bulles dans certains tissus et dans le réseau veineux qui les draine. Leur taille, mesurée
par échographie, va de 10 à 500 µm de diamètre. Il existe des bulles extravasculaires et des bulles
intravasculaires, circulantes ou non (2). Leurs effets, compressifs et hémodynamiques, apparaissent entre
quelques minutes et quelques heures après l’émersion.
Les effets compressifs sont surtout le fait des bulles extravasculaires. Ils s’observent au niveau :
- de la peau, avec compressions irritatives des terminaisons nerveuses proprioceptives ;
- des tendons, capsules articulaires et extrémités osseuses, sous la forme de douleurs périarticulaires (les
"bends") en réponse à la distension mécanique ;
- des liquides de l’oreille interne, avec destruction des structures sensorielles .
Les bulles veineuses ,créant des ischiémies veineuses ,sont drainées vers les cavités droites du cœur et
embolisent la circulation pulmonaire .avec hypertension artérielle pulmonaire et réduction du débit de
l’artère pulmonaire. Ce ralentissement se répercute en amont dans le système azygos et lombaire
ascendant, entraîne une stase veineuse des plexus lombaires, extra et périduraux. et est responsable des
formes neurologiques médullaires (3),avec lésions étagées des faisceaux de la moelle .Les bulles
artérielles présentes dans l’oreillette droite peuvent, à l’occasion d’une variation de pression
intrathoracique, franchir le filtre pulmonaire ou un foramen ovale perméable et d’emboliser la circulation
artérielle. créant des ischémies cérébrales, cochléovestibulaires ou médullaires cervicales.
I- 2. La maladie de la décompression
Les bulles sont ubiquitaires. Elles s'infiltrent dans les réseaux capillaires pulmonaires, d’où elles sont
normalement éliminées, ou systémiques, où elles exercent leurs effets délétères. Elles sont en contact avec

le contenu (plasma et éléments figurés) et le contenant (endothélium)
I- 2.1. Interactions bulles – paroi
I- 2.1.1. Au niveau de la lame basale
Les bulles ont un effet abrasif sur l'endothélium (4), mettant à nu le collagène de la lame basale, dont les
protéines (laminines, fibronectines, collagènes, facteur de von Willebrand) sont reconnues par des
intégrines plaquettaires (glycoprotéines ), entraînant l’adhésion des plaquettes au sous-endothélium et leur
activation.
I- 2.1.2. Activation des cellules endothéliales
Lorsqu’en raison de l’arrêt circulatoire les protéoglycans transmembranaires de la cellule endothéliale ne
sont plus soumis au flux circulatoire, ou que ses récepteurs reconnaissent des molécules impliquées dans
les processus de coagulation (thrombine) ou de l’inflammation (cytokines, IL-1, TNFα), la cellule
endothéliale s’active et devient thrombogène. Elle exprime à sa surface le facteur V,et de nombreux
facteurs procoagulants y compris le facteur de Willebrand , et le PAF (Platelet Activating Factor).Cette
activation entraîne un déséquilibre dans la sécrétion des médiateurs de la vasomotricité. Lors de
l’inflammation, ce déséquilibre est en faveur de la vasodilatation précapillaire Dans l’ischémie-
reperfusion ce déséquilibre est inversé au profit des EDCF (Endothelium Dependent Contracting Factors)
: prostaglandines, radicaux libres (anion superoxyde) et endothélines (ET-1, 2 et 3). Les ETont un pouvoir
vasoconstricteur extrêmement élevé et une durée de vie de plusieurs minutes (vasospasmes de longue
durée). Or, Le poumon semble être l’organe-clé dans la production d’ET-1 circulante (5),dont le rôle est
évoqué dans le développement des lésions neurologiques de la moelle épinière après traumatisme et est
établi dans l’hypertension artérielle pulmonaire de l’hypoxie.
II- 2.2. Interactions bulles - plasma
A l'interface gaz-bulle se produit un réarrangement des macromolécules entraînant une modification de
l’équilibre des charges électriques, avec création d’une couche électronégative capable d’activer le
système de contact : le facteur XII (facteur de Hagemann) s’adsorbe sur cette surface et active du facteur
XI, du facteur VII (première étape de la coagulation), du plasminogène (précurseur de la plasmine,
enzyme centrale de la fibrinolyse) et du système du complément. Des élévation de la concentration des
fractions C3a, C4a et C5a ont été mesurées après bullage in vitro dans du sang complet et retrouvées in
vivo.L’activation de ce système pré-coagulant rend disponible le facteurs VIIa qui,en présence de facteur
tissulaire (normalement non exprimé à la surface vasculaire de l’endothélium), conduit à la
fibrinoformation.
I- 2.3. Interactions avec les éléments figurés du sang.
I- 2.3.1. Les plaquettes.
Des variations du taux de plaquettes circulantes ont été rapportées dès 1969 dans les phénomènes de
 6
7
8
9
10
11
12
13
14
15
6
7
8
9
10
11
12
13
14
15
1
/
15
100%