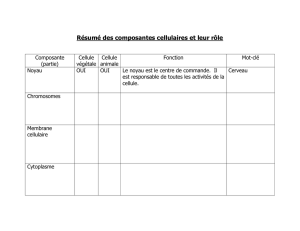
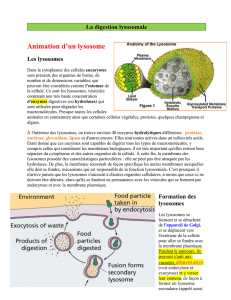
Les lysosomes


Le lysosome fut décrit et nommé en 1955 par
Le lysosome fut décrit et nommé en 1955 par
Christian René de Duve (dans les cellules de
Christian René de Duve (dans les cellules de
foie)
foie).
.
Il l’a décrit comme un organite cellulaire ,
Il l’a décrit comme un organite cellulaire ,
riche en enzymes des hydroladses
riche en enzymes des hydroladses
(enzymes hydrolytiques, lysantes) qui sont
(enzymes hydrolytiques, lysantes) qui sont
utilisées pour dégrader les macromolécules.
utilisées pour dégrader les macromolécules.
Ce sont des sacs membraneux présents dans le
Ce sont des sacs membraneux présents dans le
cytoplasme des cellules eucaryotes mise à part
cytoplasme des cellules eucaryotes mise à part
les hématies
les hématies
.
.
Ils peuvent être considérés comme l’estomac
Ils peuvent être considérés comme l’estomac
de la cellule.
de la cellule.

!""#
!""#
$%
$%
&
&
'(
'(
()'(
()'($
$()*
()*
'(
'(+
+()*
()*,
,
-./
-./
&%
&%
0!1/
0!1/
%
%

Les enzymes hydrolytiques et la
Les enzymes hydrolytiques et la
membrane lysosomiale sont produites par
membrane lysosomiale sont produites par
le
le REG
REG
l’appareil de golgi
l’appareil de golgi (où leur
(où leur
traitement se poursuit).
traitement se poursuit).
Les lysosomes utilisent environ
Les lysosomes utilisent environ 40
40 types
types
différents
différents d’enzymes hydrolytiques
d’enzymes hydrolytiques
(protéase, nucléase, glycosidase, lipase et
(protéase, nucléase, glycosidase, lipase et
d’autre encore).
d’autre encore).
Elles sont toutes
Elles sont toutes actives
actives dans un milieu
dans un milieu
très
très acide
acide.
.
Ces organites sont capables de détruire
Ces organites sont capables de détruire
tous les constituants cellulaires
tous les constituants cellulaires
(«9
(«9suicide
suicide9des organites cellulaires9»).
9des organites cellulaires9»).

La participation active des organites au
La participation active des organites au
métabolisme entraîne leur altération.
métabolisme entraîne leur altération.
Il est important dans les cellules
Il est important dans les cellules
pérennes (les cellules nerveuses) que ces
pérennes (les cellules nerveuses) que ces
éléments puissent être éliminés.
éléments puissent être éliminés.
Une propriété importante des lysosomes
Une propriété importante des lysosomes
est leurs stabilité dans la cellule vivante.
est leurs stabilité dans la cellule vivante.
Le concept des lysosomes est à l’origine
Le concept des lysosomes est à l’origine
un concept biochimique:
un concept biochimique:
Il provient du développement des
Il provient du développement des
techniques de fractionnement cellulaire
techniques de fractionnement cellulaire
par centrifugation.
par centrifugation.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
1
/
27
100%