Travaux pratiques d`immunologie

Travauxpratiquesd’immunologie
I. Introduction
Cesmanipulationsontpourbutd’étudiercertainestechniquesimmunologiques,decomprendreles
mécanismesmoléculairesmisenjeudanschacune,d’entirerl’intérêtetdepossiblesapplicationset
decomprendrealorslapuissancedecettesciencequiestaujourd’huienpleineexpansion.
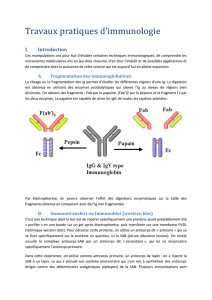
A. Fragmentationdesimmunoglobulines
LeclivageoulafragmentationdesIgpermetd’étudierlesdifférentesrégionsd’uneIg.Ladigestion
estobtenueenutilisantdesenzymesprotéolytiquesquicliventl’Igauniveauderégionsbien
distinctes.Onobtientdesfragments:Fabparlapapaïne,(Fab’)2parlapepsineetlefragmentFcpar
lesdeuxenzymes.LapapaïneestcapabledecliverlesIgGdetouteslesespècesanimales.
Parélectrophorèse,onpourraobserverl’effetdesdigestionsenzymatiquessurlatailledes
fragmentsobtenusencomparantavecdel’Ignonfragmentée.
B. ImmunotransfertouImmunoblot(westernblot)
C’estunetechniquedontlebutestderepérerspécifiquementuneprotéineayantpréalablementété
«purifier»enunebandesurungelaprèsélectrophorèse,puistransféréesurunemembranePVDL
(techniquewesternblot).Pourdétectercetteprotéine,onutiliseunanticorpsdit«primaire»quiva
sefixerspécifiquementsurlaprotéineenquestion,icilaSAB(sérumalbuminebovine).Onrévèle
ensuitelecomplexeanticorps‐SABparunanticorpsdit«secondaire»,quiluivareconnaitre
spécifiquementl’anticorpsprimaire.
Danscetteexpérience,onutilisecommeanticorpsprimaire,unanticorpsdelapin:onainjectéla
SABàunlapin,cequiastimulésonsystèmeimmunitairequis’estmisàsynthétiserdesanticorps
dirigéscontredesdéterminantsantigéniques(épitopes)delaSAB.Plusieursimmunisationssont

nécessaires(systèmederappel).Onpourrapréleverensuitelesangdulapin,lelaissercoaguler,et
enrécupérerlesérum.Onestdoncenprésenced’unsérumdelapinanti‐SAB:c’estcequ’onutilise
danscetteexpérience.
L’anticorpssecondaireestobtenuparimmunisationdechèvrecontrel’anticorpsdelapinantiSAB,
selonunprotocolesimilaire.Onycoupleensuitelaphosphatasealcalinequipermettralarévélation.
C. ELISA(EnzymeLinkedImmunoAssay)
Cettetechniqueimmunologiquetrèsutiliséeenmédecine,estutilisépourdétecterletitred’un
anticorpspourunantigènedonnéetdoncd’évaluersonaffinitérelative.Onpeutégalementétudier
leshomologiesetlesinteractionsentredifférentsantigènes.
Onétudiecilaréactivitédusérumpolyclonaldelapinanti‐SABvis‐à‐visdelaSABetdeL’OVA
(Ovalbumine).NotrebutdansceTPestdevoirs’ilestpossiblededoserdelaSABavecunanticorps
anti‐SABetdetesterlaspécificitédel’anticorps,enregardantsicelui‐cireconnaitl’ovalbumine.
II. Matérieletméthode
A. FragmentationdesIg
Lesimmunoglobulinesutiliséessontissusd’unsérumdelapin.Onréaliseunedigestion,à37°C,
pendant30minutesetpendant1h30,pourmontrerl’efficacitédelaréactionenfonctiondutemps.
DigestionàlaPapaine:LesIgsontplacéesà2mg/mLdansuntampondedigestion(PBS,0,02M
EDTA,0,02cystéine).Onenplace50µldans4tubesEppendorf.Danslestubes1et2,onmetduPBS,
danslestube3et4,onplacedelasolutionenzymatique.Lestubes1,2et3sontincubéspendant30
minutespuislaréactionestarrêtéeavecdel’iodoacétamine.Enfait,celui‐cisertseulementdansle
tube3,maisonenmetdanslestubes1et2pouravoirdessolutionscomportantlesmêmes
composés:ceciévitedesvariationsnonsignificativesdesrésultats.Letube4estincubé1h30,puisla
réactionestaussiarrêtéepardel’iodoacétamine.
DigestionàlaPepsine:Cetteexpériencefutréaliséeparl’autregroupeselonunprotocolesimilaire,
lebutrecherchéétantlemême,c'est‐à‐direunedigestiondel’immunoglobulineparl’enzyme.On
notecependantquel’onréaliselaréactionàpH=4:lapepsinepossèdeuneactivitéoptimaleàpH=
2,cependantàcepH,l’immunoglobulinenesupportepascepH.OntravailledoncàpH=4,pH
supportéparlaprotéineetpermettantuneactivitéenzymatiqueassezimportantepourunecoupure
del’Ig.
Purification:Lessolutionssontensuitedialyséestoutelanuit,à4°C,contreduPBS.Cecipermetune
purificationpartielledel’Immunoglobuline(oudesdifférentsfragmentsdel’Ig).
Onprépareensuitelegeld’acrylamide‐Bisacrylamideutilisépourlamigration.Celui‐cicomporte2
parties,legeldeconcentrationetlegeldeséparation.Legeldeconcentrationpermetunemigration
rapideetuneconcentrationdescomposésdechaquesolutionàl’interfaceentrelesdeuxgels,cequi
permetàtouslescomposésdecommencerleurmigrationdepuislemêmepoint.Legelestsuite
placédanslacuved’électrophorèsecomportantletamponadéquat.

Chaqueéchantillonestmisdansuntampondecharge,appeléLaemmli.Celui‐cipossèdeunedensité
importante,cequifaciliteledépôt,étantdonnéquel’ondéposel’échantillondanslespuitsdugel
immergé.LeBleudebromophénolqu’ilcontientestuncomposédebaspoidsmoléculairequimigre
trèsviteetquipermetdoncdesuivrelefrontdemigration.LeSDS(sodiumdodécylsulfate)confère,
auxmoléculesdelasolution,lamêmecharge,enentourantcelles‐cid’unmanteaunégatif,les
protéinesnemigrentalorsqueselonleurtaille.
Onutilisedeuxtamponsdechargedifférents:untamponréducteur(avecduβmercaptoéthanol)
pourletube2etunnonréducteurpourlestubes1,3et4.Aprèsmélangedutampondechargeet
dessolutions,onplacelestubesà95°Cpendant5minutes,pourunebonnerépartitionduSDS(grâce
àl’agitation),etuneréductiondespontsdisulfuresdesIgparleβmercaptoéthanoldansletube2.
Ondéposeensuiteleséchantillonsdanslespuitsainsiqu’untémoindemasse(TM).
Onréaliseégalementlesdépôtssuivants,selonlemêmeprotocole(tampondechargenonréducteur
+chauffage),surmêmegel:TM,Ig,SérumAlbuminebovine,ovalbumine.Cesdépôtsservirontau
westernblot(expériencedécriteplusloin).Voicil’alluredesdépôtsdugel.
345678TMFigure1:gelSDS‐Page.
1:Igréduitetdigéréàla
papaïne1h30
2:OVA
3:SAB
4:Ignonréduit
TM:témoindemasse
5:Igréduitetdigéréàla
papaïne1h30
6:Igréduitetdigéréàla
papaïnependant30minutes
7:Igréduit
8:Ignonréduit.
21TM
Onsurveillelamigration:quandlebleudebromophénol(situéauniveaudufrontdemigration)
arriveàlalimitedugel,onstoppelamigration.
Oncoupelegelendeux,laissantpourl’instantdecotélapartiequiservirapourlewesternblot.On
colorelapremièrepartiedugelavecdubleudeCoomassie.Celui‐cisefixedefaçonaspécifiquesur
touteslesprotéines.C’estuncolorantsouventutilisépourcegenredemanipulations.Onrincelegel
àl’eaudistillépuisonpasseaumicroondepourenleverlesurplusdecolorantetalorsrévélerles
différentesbandessurlegel.
B. ImmunotransfertouImmunoblot(westernblot)
Lapremièrepartiedecetteexpérienceestl’électrophorèseexpliquéedansleparagrapheprécédent.
Ondoit,avantdecommencerletransfert,apprêterlegel,lamembraneainsiquelesfiltresutilisés.
OnplacelegeldansdutampondetransfertsanséthanolpourenleverleSDS.Lamembraneest
placéedansl’éthanol,puisdansletampondetransfertcontenantdel’éthanol.Lesfiltresduhauts

sontplacésdansletampondetransfertsanséthanol,etlesfiltresdubasdansletampondetransfert
avecdel’éthanol.LePlateauanodiqueestluilargementhumidifiédansdutampondetransfert
contenantdel’éthanol.Toutlesconstituantsnécessaireauwesternblotsontéquilibrésdansla
solutiondetransfert(avecousanséthanol)pourpermettreunbontransfert(pasdechangement
brusqued’environnementlorsdutransfertdugelàlamembrane).
Pôlenégatif
Sensde FiltresGel
migrationMembranePVUL
PôlepositifTampondetransfert+éthanol
Schémadu«sandwich»demigration
Lesprotéinesvontmigreretseretrouverencontactavecl’éthanolauniveaudelamembrane.Ceci
valesfaireprécipitersurlamembrane.L’éthanolestainsirépartidansledispositifpourévitertoute
précipitationavantlamembrane.
Aprèsmigration,onrécupèrelamembrane(onfaitunemarquedistinctivepourrepérerlecôtéoù
sontlesprotéines)puisonvasaturerlessitesdefixationnonspécifiquesdesprotéines(charges
résiduellesparexemple)avecdesprotéinesdelait(ontrempedansdulait).Sionmettait
directementl’Ig,celle‐cipourraits’adsorberàlamembraneendessitesnonspécifiquesn’importeoù
surlamembrane.Lesprotéinesdelaitvontsaturerlamembraneetlessitesdefixationnon
spécifique.Parceprocédé,lesIgnesefixeront(enthéorie)quedemanièrespécifiqueauxépitopes
qu’ellesreconnaitront.OnlaveensuitelesurplusdelaitavecduPBS.
Onvaensuiteréaliserl’immunodétection:Onincubependant1heureavecl’anticorps«primaire»
(lapin)dirigécontrelaSAB.Onafixationspécifiquedesanticorpssurlesprotéines(normalementsur
laSAB).
Remarque:dansunsoucid’économie,carlesanticorpssontdesmoléculestrèscoûteuses,onutilise
quetrèspeud’anticorps:onplace1mLd’unesolutionà1/200surduParafilm,quel’onrecouvre
aveclamembrane(ilfautfaireattentionausensdelamembrane:eneffetlesprotéinesnesontque
d’uncôtédelamembrane).
OnlaveensuiteavecduPBS,Tween0,1%.LeTweenestundétergentquivapermettreunemeilleure
évacuationdesanticorpsnonfixésendessitesspécifiques(l’interactionesttrèsforte,letweenne
peutpasdéplacerlesanticorpsfixésspécifiquement).OnlaveauPBSpouréliminerletween.
Onincubeavecl’anticorpssecondaire(chèvreantilapin)dirigécontrel’immunoglobulinedelapin
antiSAB,flanquéd’uneactivitéenzymatiquephosphatasealcaline.Celui‐cisefixespécifiquementsur
sacible.OnpourraensuiterinceravecduPBS,TweenetduPBScommeprécédemmentpour
éliminerl’excèsd’anticorps.
L’activitéenzymatiquedelaphosphatasealcalineestensuiterévéléeparunesolutioncommerciale
derévélation(trèscouteuseaussi,onenutilisedonclemoinspossible).Théoriquement,desbandes
decouleurapparaitrontauxendroitsdefixationdesanticorpssecondaires.Pouréviterune

colorationexcessivequiempêcheraitunebonnelecturedesrésultats,onstoppelaréactionavecde
l’eaudistillée.
C. ELISA
L’antigèneestadsorbésurlesparoisdespuitsd’uneplaquede96puits.OnplacelaSABoul’OVA
dansdutamponCarbonate‐bicarbonate,cequivapermettrel’adhésion:àpHalcalin,laSAB(ou
l’OVA)vasefixeraufonddespuits.L’antigènenonfixéestéliminéparrinçage.Ci‐dessous,la
répartitiondesantigènesdanslaplaquedemicrotitration.
Onmetlesantigènesàuneconcentrationde100µg/mL.
1234567891011
ASAB SABSABSAB SAB SAB SAB SAB SAB SABSAB
BOVA OVAOVAOVA OVA OVA OVA OVA OVA OVAOVA
CSAB SABSABSAB SAB SAB SAB SAB SAB SABSAB
DOVA OVAOVAOVA OVA OVA OVA OVA OVA OVAOVA
ESAB SABSABSAB SAB SAB SAB
FSAB SABSABSAB SAB SAB SAB
Onsatureensuiteleplastiqueavecdulait:lesprotéinesdelaitvonts’yfixerdefaçonnonspécifique
etlesanticorpsajoutésparlasuitenesefixerontquedemanièrespécifiquesurlaSAB.Onrince
l’excèsdelait.
Onajouteensuiteunesolutiond’anticorpsprimaires(sérumanti‐SAB)àtestée,àdifférentes
concentration,cequivapermettrededéterminerletitredel’anticorps.Lesdifférentes
concentrationssonteffectuéesparunesériededilutionencascade.L’excèsestensuiteéliminépar
lavage.
Ci‐dessous,l’alluredelaplaquedemicrotitrationaveclesconcentrationsdesérumdelapinpour
chaquepuits(enµg/mL).PourleslignesAetBonutilisedusérumdelapinimmunisécontrelaSAB.
PourleslignesDetE,onutilisedusérumdelapinnonimmunisécontrelaSAB.Cecipermetde
comparerlaréponseELISAavecles2typesdesérumetdedéduirel’actiondesanticorpscontenus
danslesérumdelapinimmunisé(estcequecesontvraimentlesanticorpsquiagissent?).Onpeut
établirunevaleurtémoingrâceauxlignesCetD.
DansleslignesEetF,onplacedifférentesconcentrationsdeSABoud’OVA:lebutestdevoirs’ilya
compétitionentrelaprotéineensolutionetcellefixerauplastique.PourlaligneE,onmetdelaSAB
etpourlaligneFonmetdel’OVA.LesconcentrationsindiquéesàlaligneEsontcelledel’antigène
fixéaufond(SAB);cellesindiquéeàlaligneFsontcelledelaprotéineensolution(SABouOVA).
1234567891011
A1,00000,50000,25000,12500,0625 0,0313 0,0156 0,0078 0,00390,00200
B1,00000,50000,25000,12500,0625 0,0313 0,0156 0,0078 0,00390,00200
C1,00000,50000,25000,12500,0625 0,0313 0,0156 0,0078 0,00390,00200
D1,00000,50000,25000,12500,0625 0,0313 0,0156 0,0078 0,00390,00200
E1/20001/20001/20001/2000 1/2000 1/2000 1/2000
F15,62531,2562,51252505000
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%