Déficit en galactose-1-phosphate uridyltransférase Diagnostic de

Galactosémie
Synonymes
Déficit en galactose-1-phosphate uridyltransférase
Déficit en GALT = galactosémie classique
Galactosémie Duarte
Résumé
La galactosémie classique est une maladie métabolique héréditaire et à début néonatal. Elle est
provoquée par un changement du matériel génétique. On en distingue deux variantes : la classique et la
Duarte. Dans le cas de la galactosémie classique, le patient ne parvient pas à dégrader le galactose (une
variété de sucre). Dans le cas de la galactosémie Duarte, la dégradation du galactose est réduite, mais
le patient ne développe aucun symptôme de la galactosémie.
Le lactose, une variété de sucre que l'on retrouve dans les produits laitiers, est, pendant la digestion,
transformé en glucose et galactose (deux autres types de sucre). Le glucose apporte au corps l'énergie
dont il a besoin. Le galactose doit encore être transformé en glucose. En cas de galactosémie, cette
dernière transformation n'a pas lieu. Le niveau de galactose dans le sang s'en trouve ainsi trop élevé.
Conséquence : un enfant atteint de galactosémie ne peut pas supporter le lactose. Cependant, le lactose
se trouve dans divers aliments, dont le lait maternel et le lait pour nourrissons.
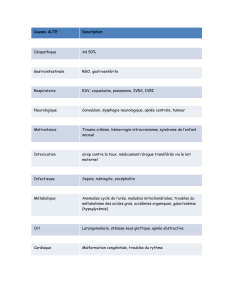
Présentation comme une septicémie peu de temps après la naissance, et en cas d'ingestion
d'aliments contenant du lactose : vomissements, léthargie, hypotonie, hypoglycémie, ictère
(cholestase, cependant également provoquée par une hémolyse), anémie hémolytique et
augmentation des transaminases jusqu'à provoquer une défaillance hépatique ;
L'évolution peut souvent se faire en une septicémie avec E. Coli pouvant donner lieu à une
méningite ;
Dysfonction tubulaire rénale (syndrome de Fanconi)
Cataracte ;
Décès immédiat ou évolution vers une cirrhose du foie, handicap mental et décompensation aiguë
des fonctions du foie lorsque la maladie n'est pas traitée ou ne l'est pas bien.
Diagnostic
La galactosémie est diagnostiquée à l'aide des symptômes susmentionnés.
Diagnostic de laboratoire métabolique : sucres réducteurs dans les urines ; galactose-1-
phosphate dans échantillon de sang (carte de dépistage néonatal*) ; diagnostic enzymatique de
GALT sur globules rouges (EDTA dans le sang ou fiche de sang néonatale). * : CAVE : la

galactosémie n'est pas décelée de manière standard dans le dépistage néonatal de masse
effectué en Flandre, mais peut être décelée de manière ciblée sur la fiche de sang.
Rarement, il s'agit d'un déficit en épimérase, qui connaît généralement une évolution clinique
moins foudroyante.
Traitement
Le traitement de la galactosémie classique consiste en un régime à vie sans galactose/lactose. Le
régime implique le remplacement notamment des produits laitiers, du pain, de la charcuterie et de
certaines variétés de fruits et légumes. Dans le cas de la galactosémie Duarte, aucun régime n'est
parfois nécessaire. Les hétérozygotes composés de la variante classique/de Duarte ont généralement
besoin de suivre un régime lors des premières années d'existence.
Incidence (fréquence)
En Belgique, la galactosémie touche environ 1 naissance sur 50.000.
Héritabilité
La galactosémie classique se transmet selon un mode autosomique récessif.
La galactosémie Duarte est également héréditaire, mais le nombre de copies du gène présentes pour la
galactosémie Duarte détermine la quantité de galactose qui n'est pas synthétisée. Si un patient ne
possède qu'une seule copie, la synthèse de galactose sera réduite d'environ 25 %. Si un patient en
possède deux copies, la synthèse de galactose sera réduite d'environ 50 %. Si une personne a une copie
du gène pour la galactosémie classique et une copie du gène pour la galactosémie Duarte, la synthèse
sera réduite d'environ 50 à 75 %.
1
/
2
100%