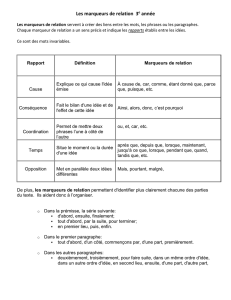
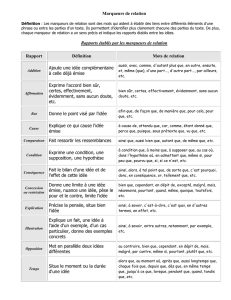
Cahier 27 - Les marqueurs cardiaques

Formation
Biologie médicale
N°27
2002
LES MARQUEURS
CARDIAQUES
CAHIER DE
Nº 27 - 2002 - CAHIER DE FORMATION BIOFORMA -
LES MARQUEURS CARDIAQUES
couv_formation27 5/11/02 13:21 Page 2

Ceci est la VERSION NUMERIQUE des CAHIERS BIOFORMA
déjà parus et distribués gratuitement* à l'ensemble des Laboratoires d’Analyses
de Biologie Médicale en FRANCE.
Ce fichier et son contenu sont la propriété de BIOFORMA.
LES DROITS D'AUTEURS SONT PROTEGES A LA B.N.F.
Toute reproduction, toute utilisation, partielle ou totale, des textes, schémas et
photos de cet ouvrage, sans l'autorisation écrite de BIOFORMA, seront
poursuivies devant les tribunaux compétents.
Seule une impression pour une copie personnelle est permise.
( étudiant, interne, biologiste de labm )
Cet ouvrage n’est pas vendu dans le commerce.
* le financement est assuré par la dotation des Caisses d’Assurance Maladie à la formation
continue conventionnelle des biologistes du secteur privé.
230 bd Raspail 75014 Paris - www.bioforma.net - [email protected]
BIOFORMA
230 bd raspail 75014 Paris

LES MARQUEURS
CARDIAQUES
Formation27.book Page 3 Mardi, 5. novembre 2002 11:56 11

LISTE DES AUTEURS
■ Dr Eric BONNEFOY
Soins intensifs / Urgences cardiologiques
Hôpital Cardiologique Louis Pradel
28, rue du Doyen Lépine
BP Lyon Montchat
69394 LYON cedex 3
Tél. : 04 72 35 71 62
Fax : 04 72 35 73 41
bonnefoy@rockfeller.uni-lyon1.fr
■ Dr Eric GARBARZ
Service de Cardiologie
Hôpital Tenon
4, rue de la Chine
75020 PARIS
Tél. : 01 56 01 67 25
Fax : 01 56 01 67 58
■ Dr M. José BUGUGNANI
Laboratoire Central de Biochimie
C.H.G. Poissy / Saint Germain en Lay
4, rue Baronne Gérard
78105 SAINT GERMAIN EN LAYE cdx
Tél. : 01 39 27 42 34
Fax : 01 39 27 42 39
Portable : 06 84 07 12 78
■ Pr Jacques INGRAND
Laboratoire de Biophysique
Faculté de Médecine Cochin
24, rue du Fg Saint Jacques
75674 PARIS cedex 14
Tél. : 01 44 41 23 75
Fax : 01 44 41 23 88
■ Dr Alain DAUNIZEAU
Service de Biochimie
C.H. du Dr Schaffner
99, route de la Bassée
Sac Postal 8
62307 LENS cedex
Tél. : 03 21 69 10 86
Fax : 03 21 69 11 84
■ Dr Guillaume LEFEVRE
Laboratoire de Biochimie
Hôpital Tenon
4, rue de la Chine
75020 PARIS
Tél. : 01 56 01 79 90
F : 01 56 01 78 40
■ Pr Jean-Yves DEVAUX
Service Médecine Nucléaire
C.H.U. Cochin
27, rue du Fg Saint Jacques
75014 PARIS
Tél. : 01 58 41 21 82
Fax : 01 58 41 21 95
■ Pr Pierre Yves MARIE
Service de Médecine Nucléaire
C.H.U. Nancy Brabois
Rue du Morvan
54511 VANDŒUVRE LES NANCY
Tél. : 03 83 15 39 09
py.marie@chu-nancy.fr
■ Pr Ludovic DROUET
Service d’Hématologie Biologique
Hôpital Lariboisière
2, rue Ambroise Paré
75010 PARIS
Tél. : 01 49 95 64 11 (sec)
Fax : 01 49 95 63 97
ludovic.drouet@lrb.ap-hop-paris.fr
■ Dr Claire RODRIGUEZ-LAFRASSE
Laboratoire de Biochimie
Hôpital Cardiologique Louis Pradel
28, rue du Doyen Lépine
BP Lyon Montchat
69394 LYON cedex 3
Tél. : 04 78 86 31 65
Portable : 06 14 94 43 14
[email protected]v-lyon1.fr
Auteurs.fm Page 4 Mardi, 5. novembre 2002 1:13 13

SOMMAIRE
5
LISTE DES AUTEURS
................................................................................................... 4
INTRODUCTION
............................................................................................................. 13
PHYSIOLOGIE CARDIAQUE (C. RODRIGUEZ-LAFRASSE)
................... 15
I. L
A
POMPE
CARDIAQUE
......................................................................................... 15
I.1-
Anatomie du cœur.............................................................................................. 15
I.2-
Système d’irrigation sanguine du cœur.............................................................. 16
I.3-
Valves cardiaques ............................................................................................... 16
I.4-
Ultrastructure...................................................................................................... 17
I.5-
Fonctionnement de la pompe cardiaque............................................................. 18
II. A
CTIVITÉ
ÉLECTRIQUE
CARDIAQUE
................................................................ 21
II.1-
Tissu de conduction et innervation cardiaque .................................................... 21
II.2-
Potentiel de repos - potentiel d’action................................................................ 22
II.3-
L’électrocardiogramme....................................................................................... 24
III. M
ÉCANISMES
DE
LA
CONTRACTION
CARDIAQUE
........................................ 28
IV. S
OURCES
D
’
ÉNERGIE
DU
MUSCLE
CARDIAQUE
............................................. 30
LES AFFECTIONS CARDIAQUES ET LEUR TRAITEMENT
................... 33
I. L
A
MALADIE
CORONAIRE
(E. B
ONNEFOY
)
.................................................... 33
I.1-
Introduction........................................................................................................ 33
I.2-
Rappels physiologiques sommaires.................................................................... 34
I.3-
L’angor stable..................................................................................................... 34
I.3.1- La maladie athéroscléreuse
.................................................................... 34
I.3.2- Description clinique
................................................................................ 35
I.3.3- L’électrocardiogramme
........................................................................... 36
I.3.4- Les techniques d’imagerie
....................................................................... 38
I.3.5- Le traitement de l’angor stable
............................................................... 39
I.4-
Les syndromes coronariens aigus....................................................................... 41
I.4.1- Physiopathologie
..................................................................................... 42
I.4.2- Les syndromes coronariens aigus sans sus-décalage du segment ST
..... 44
I.4.3- Les syndromes coronariens aigus avec sus-décalage du segment ST
..... 46
II. L’
INSUFFISANCE
CARDIAQUE
(E. G
ARBARZ
)
................................................ 60
II.1-
Épidémiologie .................................................................................................... 60
Formation27.book Page 5 Mardi, 5. novembre 2002 11:56 11
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
1
/
255
100%