Les outils du génie génétique

Les outils du génie génétique
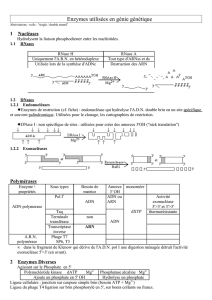
1- Les enzymes utilisées
A- Enzymes de coupure des acides nucléiques
a- Endonucléases de restriction
Ce sont des enzymes bactériennes de protection contre des ADN étrangers. On utilise beaucoup d’enzyme de
restriction de type II (coupure sur un site spécifique). On trouve deux types de coupures : à bouts francs et à bout
collants (3’-OH sortant ou 5’-P sortant). Deux enzymes différentes peuvent donner des coupures à bouts collants
compatibles. Certaines enzymes reconnaissent la même séquence, soit elles clivent de façon identiques
(isoschizomères) soit de différemment (néoisoschizomères). Certaines enzymes donnent la même coupure
(isocaudamères). Toutes les enzymes qui donnent des bouts francs sont des isocaudamères.
b- Autres nucléases
Ce sont des endo ou exonucléases (exo III : 3’ 5’ ; exo λ : 5’ 3’). Elles hydrolysent les ADN, les ARN ou les deux.
Leur coupure se fait avant ou après le phosphate. Il peut y avoir une spécificité pour les ADN simples (Mung Bean
nucléase) ou doubles brins. On trouve également les RNases H (ARN hybridés à de l’ADN).
c- Phosphatases
Ce sont des enzymes qui éliminent le phosphate en 5’ des acides nucléiques (exemple : phosphatase alcaline e
l’intestin de veau). Cela permet d’empêcher la fermeture prématurée d’un ADN circulaire et permet un marquage
radioactif par une polynucléotide kinase.
B- Les enzymes de biosynthèse
a- Les polymérases
Schéma général de la polymérisation :
Les polymérases sont des enzymes qui
copient de l’ADN ou de l’ARN (toujours
dans le sens 5’-P 3’-OH) de manière antiparallèle et complémentaire à la matrice en utilisant des nucléotide
triphospate (ARN) ou des desoxynucléotides triphosphates (ADN).
Dans le tableau ci-dessous sont cités quelques exemples :
Nécessite une matrice ADN
Nécessite une matrice
ARN
Ne nécessite pas de
matrice
ADN polymérases
DNA pol. I (exo : 5’ 3’ et 3’5’)
Klenow pol. (exo : 3’5’)
Taq pol. (exo : 3’5’)
T4 pol. (exo: ∅) (term. Transférase)
Transcriptase inverse des
rétrovirus (RNase H)
Terminale transférase
(permet de faire une
queue holopolymérique)
ARN polymérases
T7 RNA pol.
T3 RNA pol.
SPG
β Q réplicase (présente
chez des phages à ARN -)
Poly A polymérase
Les polymérases peuvent présenter d’autres activités en plus de la polymérisation : on peut trouver des activités
exonucléasiques (aussi appelées activités d’édition) 5’ 3’ (hydrolyse le dernier nucléotide incorporé s’il n’est pas
complémentaire à la base de la matrice) et 3’ 5’ (digère les amorces et permet un marquage par la technique de
nick translation). La transcriptase inverse possède une activité RNase H qui permet de détruire la matrice (qui est le
matériel génétique du virus) après la rétrotranscription. Les terminales transférases ajoutent des nucléotides en 3’
sans matrice (la Taq pol. A une activité terminale transférase pour un seul nucléotide, souvent A), ces enzymes sont
utilisées dans le processus de mutagénèse dirigée.

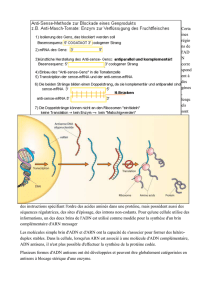
b- Les ligases, des enzymes énergie dépendantes
Elles permettent de lier des brins d’ADN ou d’ARN (si l’extrémité
5’ est phosphorylée) en utilisant l’énergie d’hydrolyse d’un ATP.
T4 RNA ligase : lie deux ADN ou ARN simples brins.
c- Polynucléotides kinases
La T4 polynucléotide kinase transfère le phosphate γ d’un ATP sur une extrémité 5’ déphosphorylée (si le phosphate
transféré est radioactif alors on a marqué l’ADN radioactivement).
2- Les vecteurs
A- Les vecteurs plasmidiques
ADN circulaire qui possède une origine de réplication qui permet une réplication autonome. Il doit porter une un
phénotype facile à repérer qui contient un site de clonage multiple. L’insert fait entre 10 et 20 kb.
a- Le plasmide pBR322 (deuxième génération : identification négative)
Il mesure 4,4 kb et possède deux gènes de résistance : tet® (pour une pompe à tétracycline) et amp® (contre
l’ampicilline). Ils ont tous les deux un promoteur. On sélectionne les recombinants grâce à des tests antibiotiques.
Cela nécessite une réplique sur velours.
b- Le plasmide pUC18 (troisième génération : identification positive)
Il mesure 2,7 kb. C’est une recombinaison entre pBR322 et le phage M13. Il possède un gène de résistance amp® et
un gène lacz’ (codant pour une protéine partielle de la β-galactosidase) contenant un site multiple de clonage. La
cellule hôte code pour l’autre partie de la β-galactosidase (E. coli ∆M15) on parle d’α-complémentation. En présence
de X-gal, les bactéries qui expriment la β-galactosidase sont bleue, les autres blanches (système blanc/bleu). On
sélectionne les colonies recombinantes sur ampicilline + X-gal + IPTG. Les colonies recombinantes sont blanches.
c- Un plasmide Eucaryote : 2μ
C’est un plasmide naturel de levure, présent à raison de 12 exemplaires par cellule (on peut en faire un vecteur
navette). Il possède un site de clonage multiple et une origine de réplication de chromosome de levure.
d- Plasmide suicides
Ce sont des plasmides qui contiennent des gènes suicides : les recombinants n’expriment pas le gène, ce sont les
seuls à pousser. Comme le gène CCdB qui code pour un inhibiteur des gyrases.
B- Vecteurs phagiques et cosmides
a- Le phage λ et ses dérivés
La tête contient 50 kb d’ADN qui se circularise dans le cytoplasme de la bactérie grâce aux séquences COS présentes
aux extrémités 5’. Dans la partie centrale du génome il y a 15 kb que l’on peut remplacer. On repère les bactéries
transformées par la radioactivité des plages de lyse. On crée des dérivés : λGT11 qui possède le promoteur de
l’opéron lactose et lacz’. Les non recombinants donnent des plages de lyse bleues.

b- Les phages à simple brin
Dérivés du phage M13 (phage à membrane) qui contient un ADNsb de 6,4 kb. Le virus ne s’exprime qu’en présence
d’un chromosome surnuméraire. La synthèse d’ADNc (complémentaire) se fait selon le schéma du cercle roulant.
L’ADN final est entouré d’une membrane, il est sécrété par fusion des membranes.
c- Les cosmides
C’est un plasmide qui a les séquences COS du phage λ, il n’y a pas de plage de lyse.
C- Les YAC
Il s’agit de chromosomes artificiels de levure. Il possède des télomères et permet une réplication sous forme linéaire
on peut cloner jusqu’à 500 kb.
3- Les sondes
A- Les sondes nucléiques
a- Obtention d’une sonde
Par synthèse chimique si on connait la séquence. Si on ca la protéine on peut faire des guessmers (10 bases).
A partir d’ADNc (complémentaire de l’ARNm).
A partir d’ARNm (en théorie).
b- L’hybridation moléculaire avec la sonde
Il faut des conditions de stringence particulières. La double hélice d’ADN présente des répulsions électrostatiques qui
sont stabilisées en présence de sels. La présence de formamide ou d’urée ainsi que la température joue sur les
conditions de stringence. Une forte stringence augmente la spécificité. Plus la sonde a un CG% élevé, plus la Tm est
élevée. Il faut que l’hybridation se fasse à une température proche de la Tm pour une meilleure spécificité.
c- Marquage des sondes
En 5’ par la T4 polynucléotide kinase avec de l’ATP radioactif en γ (mieux si déphophorylé par phosphatase
alcaline avant).
En 3’ par la terminale transférase avec un ATP marqué.
Marquage par « nick translation » après traitement à la DNase I, avec une polymérase (ADN pol. I) et des
nucléotides marqués.
Marquage par « random priming » après chauffage, on hybride des hexamères de façon aléatoire sur l’ADN
et on polymérise à partir de ces amorces avec des nucléotides marqué.
B- L’utilisation pratique des sondes
a- Southern blot et Northern blot (resp. pour ADN et ARN)
Après électrophorèse de fragment de restriction, on transfère les acides nucléiques sur une membrane par
capillarité ou électrotransfert. On chauffe la membrane pour mieux fixer les acides nucléiques. Afin d’éviter
l’adsorption non spécifique de sonde on fait une préhybridation avec des acides nucléiques de séquence différente
de la sonde. On immerge ensuite pour l’hybridation spécifique, on lave et on révèle l’emplacement de la sonde.
b- Les puces à ADN (voir cours sur les puces)

4- Les méthodes de clonage
A- Clonage classique et clonage par déphosphorylation
On place le plasmide ouvert et l’insert ensemble, on fait une ligation suivie d’une transformation. On étale les
bactéries sur un milieu sélectif ou discriminant.
Pour le clonage par déphosphorylation on met en contact l’insert et le plasmide ouvert déphosphorylé. Après
ligation les plasmides recombinants sont circulaires (mais comporte deux ruptures). Ils sont les seuls à pouvoir faire
la transformation, ils sont réparés in vivo.
B- Compatibilité des extrémités
a- Extrémités franches
Toutes les extrémités franches sont compatibles entre elles. On peut les rentre collantes grâce à des terminales
transférases qui forment des queues homopolymériques (on met un seul dNTP dans le mélange réactionnel). On
arrête la réaction au bout de quelques secondes. Il y a appariement même si la queue ne fait pas la même longueur.
On peut faire des queues avec la Taq pol. qui va donner une extrémité 3’ sortante avec un A.
b- Extrémités cohésives
Les extrémités cohésives entre elles formées par des isocaudamères sont compatibles.
c- Extrémités collantes 3’ sortantes
On va réaliser un polissage pour les transformer en extrémités franches à l’aide de différentes enzymes : soit la
Mung Bean nucléase, soit les polymérases à activité exonucléasique 3’ 5’ avec les dNTP.
d- Extrémités collantes 5’ sortantes
T4 pol. + dATP + dGTP
T4 pol. + dCTP + dTTP
3
5

C- Adaptateurs et linkers
D- PCR avec primer-linker
Si on doit pas de sites compatibles entre l’insert et le vecteur, on peut faire une PCR avec des amorces dont
l’extrémité 5’ forme un linker qui pourra être digéré après la PCR pour permettre l’insertion du fragment d’ADN.
E- Orientation du clonage
On fait de part et d’autre de l’insert des extrémités non cohésives. On va pouvoir orienter le clonage en coupant le
vecteur à l’aide d’enzymes qui donneront les extrémités complémentaires de l’insert. Il y a délétion d’un fragment
du vecteur.
1
/
5
100%