Lire l`article complet

La Lettre du Neurologue - n° 2 - vol. IV - avril 2000 105
MISE AU POINT
PHYSIOPATHOLOGIE
Le syndrome myasthénique de Lambert et Eaton (SMLE) résul-
te d’une altération préjonctionnelle de la transmission choliner-
gique au niveau des jonctions neuromusculaires (1).
L’étiopathogénie du SMLE est auto-immune, comme le suggè-
rent l’association fréquente avec d’autres pathologies auto-
immunes et l’efficacité clinique des traitements immunosup-
presseurs (2). Les caractéristiques électrophysiologiques du
SMLE sont reproduites chez l’animal par le transfert passif
d’IgG de patients atteints, impliquant leur responsabilité dans
cette pathologie (1). La cible antigénique responsable du SMLE
est probablement un canal calcique voltage-dépendant (VGCC
pour “voltage-gated calcium channel”) présynaptique. La pré-
sence chez les patients d’anticorps dirigés contre ces canaux
entraîne une réduction du nombre de canaux fonctionnels et
diminue la libération d’acétylcholine par la terminaison nerveu-
se, ce qui oppose le SMLE à la myasthénie, où le dysfonction-
nement est postsynaptique par blocage du récepteur à Ach situé
sur les cellules musculaires (1).
CLINIQUE
L’âge moyen de début du SMLE est d’environ 54 ans, avec une
légère prédominance masculine. La symptomatologie du SMLE
résulte de la double atteinte des plaques motrices neuro-musculaires
et du système végétatif cholinergique. Le début des symptomes
est habituellement insidieux. La fatigabilité musculaire en est le
signe cardinal, constamment retrouvée lors de l’évolution. Elle
prédomine aux membres inférieurs et au niveau proximal,
entraînant un handicap à la marche, à la montée des escaliers ou
au lever d'un siège. Certains patients se plaignent de myalgies à
l’effort. Les réflexes ostéo-tendineux sont faibles ou absents,
pouvant faire évoquer à tort une neuropathie périphérique.
L’augmentation de la force musculaire ou la réapparition d’un
réflexe ostéo-tendineux après un effort musculaire soutenu de
quelques secondes réalise le phénomène dit de “facilitation” ou
de “potentiation”, très évocateur du diagnostic de SMLE.
L’atteinte des paires crâniennes, retrouvée chez 70 % des
patients, est discrète et souvent fluctuante, réalisant générale-
ment une diplopie (50 %) ou un ptosis (40 %). L’interrogatoire
doit rechercher des signes d’atteinte végétative (80 % des
patients) se manifestant surtout sous la forme d’une sécheresse
buccale (75 %) ou d’une impuissance chez l’homme (45 %).
Ainsi, le SMLE se distingue cliniquement d’une myasthénie par
l’atteinte végétative et le phénomène de facilitation postexerci-
ce. Le test au Tensilon (éphédrine), souvent positif, ne permet
pas de différencier un SMLE d’une myasthénie (3, 4).
ÉLECTROMYOGRAMME
L’étude électrophysiologique est l’examen paraclinique indis-
pensable pour confirmer le diagnostic (5). Il montre des signes
spécifiques d’altération présynaptique de la transmission neuro-
musculaire, se caractérisant par l’association :
Le syndrome de Lambert-Eaton
● A.F. Carpentier, N. Le Forestier, J.Y. Delattre*
■ Le syndrome myasthénique de Lambert-Eaton (SMLE)
est une pathologie auto-immune de la jonction neuromus-
culaire qui associe fatigabilité musculaire et signes de dys-
autonomie.
■ L’électromyogramme se caractérise par des potentiels
d’action moteurs d’amplitudes réduites au repos et augmen-
tées lors d’une stimulation supramaximale à haute fréquen-
ce (>10 Hz).
■ Des anticorps dirigés contre les canaux calciques volta-
ge-dépendants présynaptiques sont retrouvés chez plus de
90 % des patients.
■ Le SMLE est d’origine paranéoplasique dans 60 % des
cas, le cancer responsable étant généralement un cancer du
poumon à petites cellules.
■Outre le traitement du cancer, la prise en charge du
SMLE repose sur les traitements immunosuppresseurs,
les plasmaphérèses, les immunoglobulines intraveineuses
ou la 3,4-diaminopyridine.
POINTS FORTS
POINTS FORTS
* Fédération de neurologie Mazarin, hôpital de la Salpêtrière, Paris.
0040027 Maq+couv ok 06/08/02 14:17 Page 105

MISE AU POINT
La Lettre du Neurologue - n° 2 - vol. IV - avril 2000
106
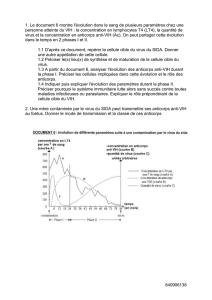
•d’une amplitude réduite des réponses motrices (souvent infé-
rieures à 1 ou 2 mV) dès la première stimulation ;
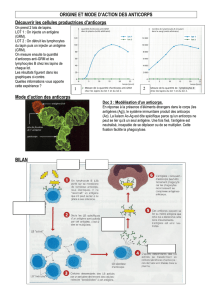
•d’une augmentation franche de l’amplitude et de la surface des
réponses motrices après stimulation répétitive à haute fréquen-
ce (20 Hz) (phénomène de potentiation avec un incrément supé-
rieur à 50 %, le plus souvent supérieur à 200 %) et/ou après une
contraction soutenue de 15 secondes dans le muscle considéré
(facilitation postexercice avec augmentation supérieure à 50 %)
(figures 1 et 2).
Le rétablissement d’une amplitude normale du potentiel moteur
après l’exercice ou une stimulation répétitive à 20 Hz traduit le
recrutement, par le nerf, de nombreuses fibres musculaires qui
restaient encore bloquées au repos ou à la stimulation simple.
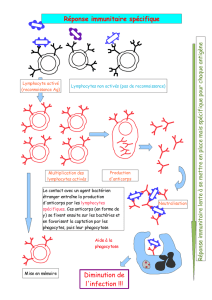
À ces signes spécifiques s’ajoutent des signes non spécifiques :
•la présence d’un décrément des potentiels moteurs lors de la
stimulation à basse fréquence (3 Hz), comme on peut le voir
dans la myasthénie (figure 3) ;
•l’existence de potentiels d’unité motrice brefs ou indentés lors de
l’exploration à l’aiguille en EMG dans le muscle.
Une atteinte dysautonomique doit être recherchée systématiquement.
Les vitesses de conduction sensitivo-motrice, les ondes F et les
réflexes H sont le plus souvent normaux, même si une polyneuropa-
thie sensitivo-motrice associée n’est pas rare pour certains auteurs (4).
ANTICORPS
La détection d’anticorps dirigés contre les canaux calciques vol-
tage-dépendants (VGCC pour “voltage-gated calcium channels”)
présynaptiques et parfois d’une grande utilité diagnostique chez
les patients peu coopérants ou chez ceux présentant un tableau
neurologique complexe. Plusieurs sous-types (T, L, N, et P/Q)
de VGCC ont été décrits, et des tests radio-immunologiques uti-
lisant des ligands spécifiques des sous-types P/Q (conotoxin-
MVIIC) et N (conotoxin-GVIA) ont été récemment développés
pour détecter des anticorps spécifiques. La détection d’anticorps
dirigés contre le sous-type P/Q (anticorps anti-P/Q) est la plus
utile en clinique, puisque ces anticorps sont trouvés chez plus de
90 % des patients avec un SMLE, alors que seulement un tiers
ont des anticorps dirigés contre le sous-type N. Si la sensibilité
à ces anticorps est bonne pour porter le diagnostic de SMLE,
leur spécificité est sujette à débat, puisque certains ont rapporté
leur présence dans d’autres pathologies neurologiques telle
la maladie de Charcot. Des études ultérieures ont cependant
contredit ces résultats, et il est raisonnable de considérer que la
détection d’anticorps anti-P/Q justifie la réalisation d’un élec-
tromyogramme à la recherche d’un SMLE non diagnostiqué ou
subclinique. Précisons enfin que le titre d’anticorps anti-P/Q
n’est pas corrélé à la gravité des symptômes cliniques ou élec-
trophysiologiques (6).
SMLE D’ORIGINE PARANÉOPLASIQUE
Près de 60 % des patients avec SMLE présentent un cancer
associé, faisant suspecter une origine paranéoplasique de leur
syndrome. Dans ce groupe de patients, les symptômes de SMLE
précèdent le diagnostic de cancer, qui est trouvé dans les deux
ans dans 95 % des cas. Si, malgré des recherches répétées, aucu-
ne tumeur n’est trouvée cinq ans après le début des symptômes
de SMLE, l’origine paranéoplasique peut être a priori écartée
(3). Le cancer associé est dans 95 % des cas intra-thoracique, et
dans plus de 85 % des cas un cancer du poumon à petites cel-
lules. Ainsi, le bilan carcinologique doit impérativement com-
porter un scanner thoracique hélicoïdal. Les autres cancers peu-
vent être, entre autres, des carcinomes pulmonaires, des cancers
de la prostate ou du sein, des thymomes, des lymphosarcomes.
La réponse immune anti-VGCC est probablement déclenchée
Figure 1. Amplitude
réduite à la première
stimulation. Facilitation
postexercice à la 2e
stimulation. (Extrait de
E. Fournier, 1998. Examen
électromyographique
et étude de la conduction
nerveuse. Sémiologie électro-
physiologique, Ed EMinter).
Figure 2. Test de stimulation répétitive à 20 Hz : potentiation de plus
de 400 %.
(Extrait de E. Fournier, 1998. Examen électromyographique et étude de la conduction ner-
veuse. Sémiologie électrophysiologique, Ed EMinter.)
Figure 3. Test de simulation
répétitive à 3 Hz : décrément
d’amplitude de 27 %.
(Extrait de E. Fournier, 1998. Examen
électromyographique et étude de la
conduction nerveuse. Sémiologie
électrophysiologique, Ed EMinter.)
0040027 Maq+couv ok 06/08/02 14:17 Page 106

La Lettre du Neurologue - n° 2 - vol. IV - avril 2000 107
par l’expression des VGCC par la tumeur, puisque les cancers
du poumon à petites cellules expriment le sous-type P/Q de
VGCC. Même si le traitement de la tumeur entraîne parfois une
rémission des symptômes du SMLE (7),le pronostic de ces
patients est sombre, avec une médiane de survie inférieure à un an,
en rapport avec une progression tumorale (3).
Le SMLE est parfois associé à d’autres syndromes paranéopla-
siques. Ainsi, 12 % des patients présentant un anticorps anti-Hu
ont aussi des anticorps anti-P/Q. Les dégénérescences cérébel-
leuses paranéoplasiques (DCP) représentent un cas particulière-
ment intéressant, puisque près d’un tiers des patients présentant
une DCP avec un cancer du poumon à petites cellules ont des
anticorps anti-P/Q (8). À l’inverse, aucune patiente avec DCP et
cancer gynécologique n’a d’anticorps anti-P/Q. Il en résulte que
la détection d’anticorps anti-VGCC chez les patients présentant
une atteinte cérébelleuse subaiguë d’étiologie indéterminée est
fortement évocatrice d’un syndrome paranéoplasique et d’un
cancer pulmonaire à petites cellules sous-jacent.
SMLE D’ORIGINE AUTO-IMMUNE
Concernant les 40 % de patients chez qui aucun cancer n’est
retrouvé, l’étiologie du SMLE est probablement auto-immune.
En effet, ces patients ont généralement des antécédents person-
nels ou familiaux de pathologie auto-immune, comme un vitili-
go, une anémie pernicieuse, une maladie cœliaque, un diabète
ou une thyroïdite auto-immune (3). Aucun critère clinique ou
électrophysiologique ne permet de distinguer l’origine paranéo-
plasique ou non paranéoplasique d’un SMLE. Les anticorps
anti-VGCC sont présents dans les deux groupes de patients.
Chez les patients dont le SMLE n’est pas d’origine paranéopla-
sique, la médiane de survie est de l’ordre de sept ans, avec des
cas de survie longue, parfois supérieure à quinze ans (3).
PRISE EN CHARGE DES SMLE
La prise en charge des SMLE bénéficie de multiples thérapeu-
tiques efficaces. Le traitement de la tumeur dans les cas paranéo-
plasiques améliore parfois la symptomatologie (7). La corticothé-
rapie ou l’azathioprine, ou idéalement l’association des deux, sont
efficaces au bout de quelques semaines (2). Les échanges plas-
matiques ou les perfusions d’immunoglobulines intraveineuses
améliorent la symptomatologie et les altérations électrophysiolo-
giques après un délai de une à trois semaines, et durent environ
deux mois (2, 9). La pyridostigmine aux doses efficaces dans la
prise en charge de myasthénie peut améliorer partiellement la
force musculaire. Parmi les agents améliorant spécifique-
ment la transmission neuro-musculaire présynaptique par bloca-
ge de la conductance membranaire (guanidine, 4-aminopyridine
ou 3,4-diaminopyridine), seule la 3,4-diaminopyridine est correc-
tement tolérée, avec des effets secondaires limités à des paresthé-
sies, des insomnies ou des effets cholinergiques et, à forte dose
(> 60 mg/j), des crises comitiales. Les effets secondaires et l’effi-
cacité clinique sont dose-dépendants, la posologie recommandée
se situant entre 20 et 50 mg par jour. Certains auteurs ont rappor-
té une accoutumance, mais la 3,4-diaminopyridine induit généra-
lement une amélioration prolongée sur plusieurs mois (10).
Malgré la multiplicité des traitements, ou peut-être à cause d’el-
le, la prise en charge des SMLE n’est pas clairement codifiée.
Dans les SMLE paranéoplasiques, le traitement de la tumeur est
impératif, et il est raisonnable de considérer un traitement par
3,4-diaminopyridine pour améliorer la symptomatologie. Dans
les SMLE d’origine auto-immune, un traitement associant pyri-
dostigmine et corticothérapie peut être proposé en première
intention, le recours en seconde intention à l’azathioprine, aux
immunoglobulines ou aux plasmaphérèses dépendant du patient
et des contingences hospitalières locales. ■
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Kim Y.I. Passively transferred Lambert-Eaton syndrome in mice receiving
purified IgG. Muscle Nerve 1986 ; 9 : 523-30.
2. Newsom-Davis J., Murray N.M. Plasma exchange and immunosuppressive
drug treatment in the Lambert-Eaton myasthenic syndrome. Neurology 1984 ;
34 : 480-5.
3. O’Neill J.H., Murray N.M., Newsom-Davis J. The Lambert-Eaton myasthenic
syndrome. A review of 50 cases. Brain 1988 ; 111 : 577-96.
4. O’Suilleabhain P., Low P.A., Lennon V.A. Autonomic dysfunction in the
Lambert-Eaton myasthenic syndrome : serologic and clinical correlates.
Neurology 1998 ; 50 : 88-93.
5. Fournier E. Examen électromyographique et étude de la conduction nerveuse.
Sémiologie électrophysiologique, Ed EMinter ; 1998 ; pp 253-84.
6. Nakao Y.K., Motomura M. et coll. Specificity of omega-conotoxin MVIIC-bin-
ding and-blocking calcium channel antibodies in Lambert-Eaton myasthenic syn-
drome. J Neurol 1999 ; 246 : 38-44.
7. Jablecki C. Lambert-Eaton myasthenic syndrome. Muscle Nerve 1984 ;
7:250-7.
8. Mason W.P., Graus F. et coll. Small-cell lung cancer, paraneoplastic cerebellar
degeneration and the Lambert-Eaton myasthenic syndrome. Brain 1997 ;
120 : 1279-300.
9. Bain P.G., Motomura M. et coll. Effects of intravenous immunoglobulin on
muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton
myasthenic syndrome. Neurology 1996 ; 47 : 678-83.
10. Lundh H., Nilsson O. et coll. Practical aspects of 3,4-diaminopyridine treat-
ment of the Lambert-Eaton myasthenic syndrome. Acta Neurol Scand 1993 ; 88 :
136-40.
0040027 Maq+couv ok 06/08/02 14:17 Page 107
1
/
3
100%