Utilisation d`un anticorps monoclonal anti-Tn en

!
"!
THÈSE DE DOCTORAT
DE L’UNIVERSITÉ PARIS 5 - RENÉ DESCARTES
École Doctorale Gc2iD - ED 157
Spécialité : Biologie cellulaire et moléculaire
Présentée par
Adèle HEITZMANN-DAVERTON
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITÉ PARIS 5
Sujet de la thèse :
Utilisation d’un anticorps monoclonal anti-Tn
en immunothérapie des cancers
Soutenue le 28 Juin 2013
à l’Institut Curie, Paris
Devant le jury composé de :
Monsieur le Docteur Sebastian AMIGORENA (DR) Directeur de thèse
Madame le Professeur Mireille VIGUIER Présidente du jury
Madame le Professeur Claude LECLERC Rapporteur
Monsieur le Docteur Pierre BRUHNS (DR) Rapporteur
Madame le Docteur Nathalie CHAPUT-GRAS Examinateur

!
#!
A mon mari David
A mes parents

!
$!
!"#"!$%"#"&'()
Ce travail de thèse a été réalisé à l’Institut Curie, au sein du laboratoire « Immunité et Cancer »
(Inserm U932) dirigé par Monsieur le Docteur Sebastian AMIGORENA.
Je souhaiterai tout d’abord remercier les membres du jury, qui ont bien voulu me faire l’honneur
de juger mon travail, et notamment la présidente de ce jury, Madame le Docteur Mireille
VIGUIER.
Je remercie plus particulièrement Madame le Docteur Claude LECLERC et Monsieur le Docteur
Pierre BRUHNS qui ont accepté la tâche d’être rapporteurs de ce manuscrit.
Un grand merci également à Madame le Docteur Nathalie CHAPUT-GRAS pour avoir accepté
d’évaluer ce travail de thèse en tant qu’examinatrice.
Mes remerciements s’adressent ensuite à Sebastian AMIGORENA, directeur de l’Unité Inserm
U932, pour m’avoir accueillie au sein de son laboratoire et pour avoir accepté d’être mon
directeur de thèse.
Je remercie également Pascale HUBERT et Christine SEDLIK, qui m’ont accueillie au sein de leur
équipe et m’ont guidée tout au long de mon projet de thèse. Pascale, je te remercie car c’est avec
toi que j’ai effectué mes premiers pas dans le monde de la recherche, sous ton œil attentif et
rigoureux. C’est avec toi que j’aurai publié mon premier article scientifique. Christine, merci
d’avoir accepté de m’encadrer suite au départ de Pascale et d’avoir repris la suite du projet Tn.
Merci aussi à Sophie VIEL, Assistante Ingénieur de notre équipe Tn, pour son aide, et pour avoir
pris soin de mes cultures cellulaires et de mes souris.
Merci également à Claire HIVROZ pour avoir accepté d’être ma tutrice de thèse, pour son écoute
et ses conseils.
Je remercie également l’équipe de Frédéric SCHMIDT du laboratoire de "Conception, Synthèse et
Vectorisation de Biomolécules" (UMR 176, Institut Curie) dirigé par Monsieur Jean-Claude
FLORENT, pour avoir accepté de collaborer avec nous dans le couplage de notre anticorps à des
molécules chimiques. Un grand merci à Rafik AIT-SARKOUH qui a réalisé lui-même tous les

!
%!
couplages avec succès, pour sa grande gentillesse, son professionnalisme, et sa détermination à
résoudre les problèmes.
J’adresse un clin d’œil à tous mes collègues de bureau « pièce 440 » : Sophie VIEL, Caroline
CHESNEAU, Jordan DENIZEAU, Philippe DE LA ROCHERE et Hélène DURET, pour leur bonne
humeur et pour tous les bons moments que nous avons passés ensemble. Caroline, tu auras été ma
grande confidente, toujours là pour me remonter le moral, écouter mes aventures campagnardes du
week-end... et ton départ pour Servier a laissé un grand vide au laboratoire. Jordan, merci pour ton
énergie débordante, ta bonne humeur et ta gentillesse. Reste comme tu es, on t’adore. Philippe, tu
es fort urbain (« du nom d’un Pape... »), toujours désagréable, mais on s’habitue... Je me rappelle
encore la première fois que je t’ai rencontré au labo, tu étais d’une humeur massacrante !
Une petite dédicace également à mes collègues de l’équipe Lantz : Héloïse FLAMENT, Virginie
PREMEL, Delphine LOUIS... et toutes les personnes de notre étage, qui chacune apporte sa
contribution à l’ambiance générale. Virginie et Héloïse, vous avez toujours été à l’écoute lors de
mes petites baisses de moral, et je vous en remercie.
J’adresse un très grand merci à ma famille, et tout particulièrement à mes parents, pour m’avoir
soutenue et encouragée pendant ces longues années d’études, avec les hauts et les bas inévitables
ponctuant toute thèse. Je serai maintenant Docteur, comme papa !
J’adresse enfin un IMMENSE MERCI à mon mari, David, car supporter sa femme en thèse, ses
multiples questionnements, ses doutes, ses baisses de moral, son mauvais caractère... n’est pas
toujours facile au quotidien... En tous cas, cela ne t’a pas empêché de me demander en mariage et
de nous unir le 1er Septembre 2012 ! Alors merci d’avoir toujours été là pour moi.

!
&!
!"(*#")
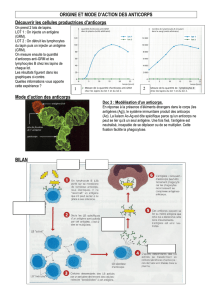
La transformation des cellules normales de l’organisme en un phénotype malin est souvent
accompagnée de changements dans leur antigénicité. L’antigène Tn (GalNac-O-Ser/Thréo) est un
antigène (Ag) glycopeptidique spécifique des tumeurs et exprimé à la membrane plasmique des
cellules cancéreuses dans la majorité des carcinomes humains ainsi que dans certaines tumeurs
hématologiques, tandis qu’il n’est pas détecté dans les cellules normales. Il représente donc une
cible potentielle très intéressante pour l’immunothérapie passive par anticorps, car il n’est pas
détectable dans les cellules normales, mais est démasqué dans environ 90% des cancers épithéliaux
du fait d’une dérégulation des processus de glycosylation.
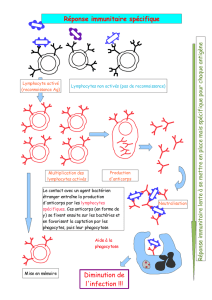
Les anticorps monoclonaux (AcM) spécifiques d’antigènes exprimés à la membrane des cellules
tumorales ont une efficacité prouvée dans le traitement de certains cancers. Ces AcM
thérapeutiques sont particulièrement intéressants pour le traitement des cancers du fait de leur forte
spécificité pour les cellules tumorales et de leur faible toxicité pour les cellules normales,
contrairement aux chimiothérapies conventionnelles, mais leur mécanisme d’action est encore mal
connu. L’AcM Chi-Tn est un anticorps chimérique homme/souris capable de se fixer de façon
spécifique à l’antigène tumoral Tn, alors qu’il ne se fixe pas sur les cellules normales. Cet AcM
pourrait donc être envisagé comme agent thérapeutique dans le traitement des cancers épithéliaux
par immunothérapie passive.
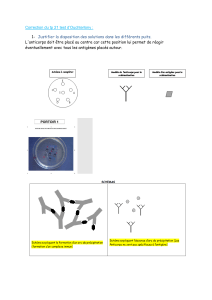
Nous nous sommes intéressés à l’AcM Chi-Tn non couplé en vue d’analyser son mécanisme
d’action et d’évaluer son efficacité thérapeutique in vivo. Nous avons montré que l’AcM Chi-Tn
seul ne possède pas d’effet toxique direct sur les lignées de cellules tumorales Tn-positives in vitro.
Cependant, en présence de macrophages, cet AcM est capable d’induire la lyse de ces cellules par
un mécanisme d’ADCC. In vivo, l’AcM Chi-Tn, associé à la cyclophosphamide, induit le rejet
d'une tumeur du sein dans plus de 80% des souris. Cette inhibition de la croissance tumorale est
abolie chez les souris déficientes pour la chaine γ associée aux récepteurs RFcγ activateurs,
suggérant in vivo un mécanisme d’ADCC. Par l’étude microscopique du microenvironnement
tumoral, nous avons observé que les cellules tumorales forment in vivo des synapses avec des
macrophages, des neutrophiles, mais aussi des lymphocytes B. Des expériences de survie in vivo
chez des souris déficientes pour différentes populations cellulaires montrent que les lymphocytes T
semblent nécessaires à la protection des souris par Chi-Tn contre la tumeur. Ainsi, ces résultats
confirment le rôle des effecteurs exprimant des RFcγ activateurs, mais aussi le rôle indispensable de
la réponse immune adaptative pour assurer l'effet thérapeutique des AcM.
Nous nous sommes également intéressés à l’utilisation potentielle de l’AcM Chi-Tn comme vecteur
d’agents cytotoxiques. In vivo, dans un modèle de tumeurs solides chez la souris, des expériences
de biodistribution montrent que l’AcM Chi-Tn est capable de cibler spécifiquement les zones
tumorales, ce qui en fait un anticorps potentiellement utilisable comme vecteur de molécules
toxiques. L’internalisation du complexe anticorps/antigène cible est un pré-requis nécessaire à
l’utilisation de l’anticorps conjugué. Nous avons montré in vitro que l’AcM Chi-Tn est internalisé
dans les endosomes précoces et de recyclage pendant un temps relativement long, faisant de cet
AcM un bon candidat pour être couplé à des agents cytotoxiques. Durant ma thèse, nous avons
couplé l’AcM Chi-Tn à la toxine saporine ou à la molécule cytotoxique auristatine F, et nous avons
montré in vitro que ces conjugués sont cytotoxiques sur des lignées cellulaires Tn-positives.
Mots clés :
Anticorps monoclonal, immunothérapie, internalisation, immunoconjugué, saporine, auristatine F.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
1
/
138
100%