La Fièvre méditerranéenne familiale : manuel du praticien

2
La Fièvre méditerranéenne familiale : manuel du praticien
La Fièvre méditerranéenne
familiale :
manuel du praticien
Salem Bouomrani
9.28 687599
----------------------------INFORMATION----------------------------
Couverture : Classique
[Roman (134x204)]
NB Pages : 104 pages
- Tranche : 2 mm + (nb pages x 0,055 mm) = 7.72
----------------------------------------------------------------------------
La Fièvre méditerranéenne familiale : manuel du praticien
Salem Bouomrani
Salem Bouomrani

2
2

2
3
Chapitre 1
Introduction/généralités
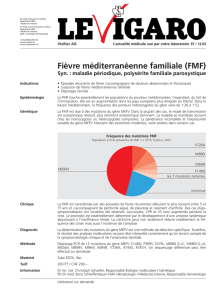
La Fièvre Méditerranéenne Familiale (FMF)
anciennement appelée « maladie périodique », est une
maladie inflammatoire chronique à évolution paroxystique
qui fut décrite pour la première fois en 1945 [1] et
caractérisée génétiquement en 1992 [2,3].
C’est une affection à caractère familial et à
transmission principalement autosomique récessive qui est
particulièrement fréquente chez les arméniens, les turques,
les juifs sépharades et les arabes du Moyen-Orient [1,4].
Son substratum génétique est une mutation ponctuelle du
gène « MEFV » situé sur le bras court du chromosome 16
[1,4, 5]. La transcription de ce gène aboutit à la production
d’une protéine baptisée « pyrine » ou « marénostrine » qui
intervient dans la régulation de la réponse inflammatoire,
en particulier leucocytaire et l’apoptose cellulaire [4,5].
Sur le plan clinique, la FMF se caractérise par des
accès paroxystiques douloureux et fébriles abdominaux,
thoraciques et articulaires auxquels peuvent s’associer
diverses manifestations systémiques : cardiaques,
neurologiques, cutanées, urogénitales et hématopoïétiques.

2
4
Son diagnostic clinique est basé sur des critères
diagnostiques dont les plus utilisés sont ceux de Livneh [6]
et sa confirmation reste du domaine de la cytogénétique
par la mise en évidence de mutations du gène « MEFV »
dont il existe plus d’une vingtaine. Les plus fréquentes de
ces mutations sont : M694V, M694I, V726A et E148Q
[4,7]. Sa complication majeure est l’amylose AA qui
domine le pronostic [1,4].
Les publications récentes font état de plusieurs
associations pathologiques possibles et d’un risque
particulièrement augmenté de dégénérescence maligne
(cancers solides et hémopathies malignes) au cours de cette
affection [8].
Sur le plan nosologique, la FMF est une fièvre
récurrente héréditaire (FRH) appartenant à la famille des
maladies auto-inflammatoires (MAI) héréditaires. Ces
fièvres récurrentes héréditaires représentent un groupe
hétérogène d’affections ayant en commun :
1. Des accès fébriles récurrents périodiques et
spontanément résolutifs associés à des manifestations
inflammatoires systémiques prédominant sur les
séreuses et les articulations.
2. Un syndrome inflammatoire biologique constant avec
une élévation permanente de la protéine de
l’inflammation SAA dont le risque majeur est une
amylose secondaire de type AA.
3. Le caractère familial avec une transmission
autosomique.
Le risque majeur de ces affections est le
développement d’une amylose secondaire type AA avec
des localisations systémiques, particulièrement rénale
pouvant mettre en jeu le pronostic vital [9-16].

2
5
Ces affections ont bénéficié ces dernières années des
progrès de la biologie moléculaires permettant leur
caractérisation génétique et la détection des différentes
mutations qui sont à leur origine, offrant ainsi un
diagnostic génétique de certitude dans les formes
douteuses ou atypiques [9-18].
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%