Chevauchement entre syndrome des antisynthétases à anti

n Revue Marocaine de Rhumatologie n
CAS CLINIQUE
Chevauchement entre syndrome des
antisynthétases à anti-PL12 et syndrome
de Gougerot-Sjögren
Overlap antisynthetase syndrome anti PL12 and Sjögren syndrome
L
e syndrome des antisynthétases
est une affection rare apparte-
nant au groupe de myopathies
inflammatoires, qui associe une
myosite auto-immune, une pneumopa-
thie interstitielle diffuse, une hyperkéra-
tose fissuraire des doigts, une polyarth-
rite, et un syndrome de Raynaud [1,2].
Il est caractérisé par la présence d’anti-
corps anti-aminoacyl-ARNt synthétases
dont les anticorps anti-PL12 qui sont
rares [3,4]. La présence de ces anticorps
peut être isolée ou associée à d’autres
anticorps dans le cadre « d’Overlap
syndrome ». En effet, il existe une pos-
sibilité de chevauchement entre le syn-
drome des antisynthétases à anti-PL12
et le syndrome de Gougerot-Sjögren.
Cette situation a été rapportée chez
23,5 % des cas selon les séries franco-
phones, avec une fréquence de surve-
nue variable [3]. Ce qui suggère que
l’association n’est pas fortuite. Nous en
rapportons une nouvelle observation.
OBSERVATION
Mme A.A. âgée de 27 ans, sans anté-
cédents pathologiques notables, notam-
ment pas d’antécédents personnels
d’endocrinopathies ni d’antécédents
familiaux de maladies auto-immunes,
qui présente depuis 4 ans une polyarth-
rite bilatérale et symétrique des grosses
et petites articulations intéressant les
mains, les poignets et les genoux. Elle
rapporte depuis 2 ans des myalgies
diffuses, une xérostomie et une
xérophtalmie, associées à une dyspnée
d’effort et un syndrome de Raynaud
depuis 1 an. Le tout évoluant dans un
contexte d’asthénie et de fièvre chiffrée
à 38-39°C. L’examen clinique a trouvé
une patiente apyrétique, une poly-
arthrite des deux poignets et des deux
genoux, une acrocyanose, une ulcération
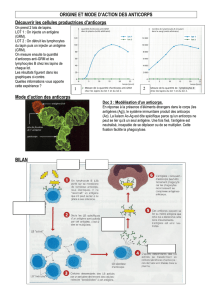
de la pulpe des doigts et orteils, et des
papules érythémateuses en regard des
métacarpo-phalangiennes (Voir Photos
1 et 2). Le bilan biologique a montré un
syndrome inflammatoire (VS = 98 mm
à la 1re heure, CRP = 59 mg/l, hyper-
gamma-globulinémie polyclonale), les
enzymes musculaires étaient normales
(CPK = 25 UI/ml, LDH = 185 UI/ml, les
GOT et Aldolase sont normales). Le bi-
lan immunologique a objectivé un taux
d’anticorps anti-nucléaires <1/80, des
anticorps anti-DNA natif négatifs, des
anticorps anti-PL12 positifs (Immuno-
dot), des anticorps anti-Ro/SSA positifs
(>240 UI/ml), et un facteur rhumatoïde
positif (Latex à 20 UI/ml, Waaler Rose
à 51,2 U/ml). Les autres anticorps anti-
synthétases notamment les anti-JO1, anti-
PL7, anti-OJ, anti-EJ, anti-JS, et anti-
KS étaient absents. Les autres anticorps
spécifiques des myosites tels que les
anti-SRP, anti-Mi-2 étaient absents. De
même que pour les anticorps associés
aux myosites recherchant une connecti-
vite associée, tels que les anticorps anti-
SSB, anti-Sm, anti-RNP (U1-U3, U5),
anti-Ku, anti-Pm-Scl, anti-SCL70, anti-
centromères et anti-CCP étaient absents.
Le bilan phosphocalcique était normal
(Ca corrigé = 94 mg/l, Ph = 43 mg/l,
PAL = 140 U/l, calciurie de 24 heures =
140 mg/24 h), ainsi que le bilan thyroï-
dien (TSH = 0,5 µUI/ml, T3 et T4 nor-
males). L’électromyogramme n’a pas
Résumé. Le syndrome des antisynthétases est un
groupe de myopathie inammatoire caractérisé par la
présence d’anticorps anti-ARNt synthétases dont les anti-
PL12 (2 %). Le chevauchement entre le syndrome des
antisynthétases à anti-PL12 et le Gougerot-Sjögren a été
rarement décrit dans la littérature. Nous en rapportons
une nouvelle observation.
Rev Mar Rhum 2012;19:45-47.
Samia Mansouri
1
, Latifa Tahiri
1
, Najoua Ghani
1
, Nadira Kadi
1
,
Samira Rabhi
2
, Wafaa Bono
2
, Taoufik Harzy
1
1- Service de Rhumatologie, CHU Hassan II - Fès / 2- Service de Médecine interne, CHU Hassan II - Fès
Syndrome des
antisynthétases, Syndrome
de Gougerot-Sjögren,
Anticorps anti-PL12,
Anticorps anti-Ro/SSA
Mots clés
n Revue Marocaine de Rhumatologie n 45

montré d’atteinte myogène. La biopsie
musculaire était normale (pas de myosite
inflammatoire). Les radiographies stan-
dards ont montré au niveau des mains un
pincement des articulations métacarpo-
phalangiennes sans érosions ni destruc-
tion articulaire. Le scanner thoracique a
montré un syndrome interstitiel bilatéral.
L’exploration fonctionnelle respiratoire,
le lavage boncho-alvéolaire, et l’écho-
graphie trans-thoracique n’ont pas mon-
tré d’atteinte pulmonaire notamment
pas de syndrome restrictif ni d’alvéolite
lymphocytaire, ni d’hypertension arté-
rielle pulmonaire. La manométrie œso-
phagienne était normale. L’examen oph-
talmologique a objectivé un syndrome
sec oculaire. La biopsie des glandes sali-
vaires accessoires a montré une sialadé-
nite stade IV de Chisholm et Masson.
Le diagnostic de syndrome des antisyn-
thétases à anticorps anti-PL12, associé à
un syndrome de Gougerot-Sjögren a été
retenu. La patiente a été mise sous pred-
nisone à raison de 1 mg/kg/j (50 mg/j)
avec dégression progressive. L’évolu-
tion a été marquée par la réapparition
des arthralgies et des myalgies à la dose
de 30 mg/j d’où la mise en route d’une
corticothérapie à dose minimale efficace
et l’adjonction du méthotrexate : 0,3
mg/Kg/semaine avec une bonne amé-
lioration clinique et biologique. Le recul
était de 2 ans.
DISCUSSION
Classiquement, le syndrome des antisyn-
thétases (SAS) associe une myopathie
inflammatoire relativement discrète, une
atteinte pulmonaire type pneumopathie
interstitielle diffuse qui pourrait condi-
tionner le pronostic de ce syndrome en
l’absence de traitement, des manifes-
tations articulaires variables allant de
simple polyarthralgie à une polyarth-
rite souvent non érosive intéressant les
mains, les poignets, les coudes et les
genoux avec parfois un facteur rhuma-
toïde positif. La présence d’un phéno-
mène de Raynaud, d’une hyperkératose
fissuraire de la pulpe et des faces latérales
des doigts donnant un aspect de « mains
de mécanicien » en caractérise l’expres-
sion clinique. Les signes généraux sont
souvent importants avec une fièvre
chez 80 % des patients, une asthénie et
un amaigrissement, avec un syndrome
inflammatoire modéré [1,5]. Sur le plan
immunologique, ce syndrome est carac-
térisé par la présence d’auto-anticorps
anti-aminoacyl-ARNt synthétases (appe-
lés communément anticorps antisynthé-
tases) dirigés contre des enzymes intra-
cytoplasmiques qui ont une fonction
cellulaire bien définie. Les patients pré-
sentant ces anticorps dits « spécifiques »
des myosites, sont classés dans la caté-
gorie des myosites de chevauchement
(Overlap myositis) définie par Troyanov
et coll., et donc potentiellement ces myo-
sites peuvent être associées à d’autres
connectivites [1,6]. Cette classification
clinico-sérologique de Troyanov permet
d’affiner le diagnostic, le traitement, et
elle a un intérêt pronostique quand à la
réponse à la prednisone et à l’évolution
chronique de cette affection. Plusieurs
spécificités d’anticorps antisynthétases
ont été identifiées notamment les anti-
JO1 qui sont les plus fréquents (20 à
30 %). Les anticorps antisynthétases
type anti-PL12, quand à eux, sont beau-
coup plus rares (<2 %), et nous dispo-
sons de peu d’études les concernant.
En effet, il existe de rares données de
la littérature concernant le SAS avec
anticorps anti-PL12, et les particularités
cliniques et biologiques de ce syndrome
sont nettement moins connues du fait
de la rareté des études réalisées dans ce
sens. Hervier et coll., ont essayé à travers
une étude rétrospective multicentrique
de comparer les manifestations cliniques
du SAS avec anticorps anti-JO1 au SAS
avec anticorps anti-PL12 [2]. Le spectre
clinique et la sévérité du SAS étaient très
variables selon les patients, aussi bien
en cas d’anticorps anti-PL12, qu’en cas
d’anticorps anti-JO1. Les fréquences re-
latives de survenue de signes généraux,
d’un phénomène de Raynaud, de mains
de mécanicien ou de symptômes articu-
laires étaient comparables dans les deux
groupes de patients. Par ailleurs, d’autres
études récentes qui se sont intéressées
aux manifestations cliniques du SAS
à anti-PL12 ont montré que ces signes
cliniques sont plus rares qu’au cours des
SAS avec anticorps anti-JO1 avec une
prévalence nettement diminuée [3,6,7].
L’atteinte musculaire au cours du SAS
à anti-PL12 varie selon les sources. Elle
est souvent rare, voire absente. Cepen-
dant lorsqu’elle est présente, elle est
peu sévère (myalgies, déficit musculaire
léger, CPK inférieur à 2 fois la normale
chez 50 % des cas seulement) [2,3,6-
8], comme est le cas de notre patiente.
L‘atteinte pulmonaire au cours de ce
syndrome est la plus constante avec une
incidence supérieure à 70 % dans les
principales études et dont la
n Revue Marocaine de Rhumatologie n
PHOTO : Ulcération de la pulpe des 2e et 3e doigts de la main gauche.
PHOTO : Papules érythémateuses en regard
des métacarpo-phalangiennes.
n Revue Marocaine de Rhumatologie n
46

n Revue Marocaine de Rhumatologie n
n Revue Marocaine de Rhumatologie n
sévérité est variable. Elle se manifeste
initialement par une toux sèche, une
dyspnée, voire un syndrome de détresse
respiratoire aiguë dans les stades avan-
cés avec fibrose pulmonaire diffuse. Le
scanner thoracique permet de mettre en
évidence une pneumopathie interstitielle
non spécifique. Les explorations fonc-
tionnelles respiratoires rechercheront
une diminution de la capacité de diffu-
sion du monoxyde de carbone (DLCO),
voire un syndrome restrictif, et le lavage
broncho-alvéolaire montrera une alvéo-
lite lymphocytaire [2,3,6,9]. Parfois, le
SAS à anti-PL12 peut se présenter sous
forme d’une pneumopathie interstitielle
non spécifique isolée (29,4 %) selon une
large série européenne [3]. De fait, le
clinicien doit être capable de rechercher
les anticorps anti-PL12 dans ce contexte.
La sévérité de l’atteinte pulmonaire et la
présence d’une hypertension artérielle
pulmonaire (complication rare de ce
syndrome) conditionnent le pronostic
du SAS à anti-PL12 et justifient une sur-
veillance des malades à la recherche de
cette atteinte pulmonaire [3,6,10,11].
Par ailleurs, la présence des anticorps
anti-PL12 au cours du SAS peut être
exclusive [3,12], ou dans certains cas,
être associée à d’autres anticorps rentrant
dans le cadre d’un « Overlap syndrome ».
Autrement dit, le SAS à anti-PL12 peut
se présenter sous la forme d’un syn-
drome de chevauchement avec d’autres
connectivites notamment le syndrome de
Gougerot-Sjögren dont le diagnostic est
basé sur des arguments cliniques, immu-
nologiques et histologiques [8,13]. Cette
situation a été observée chez 23,5 % des
cas, ce qui suggère que cette association
n’est pas fortuite, mais son mécanisme
physiopathologique reste mal connu [3].
La possibilité d’une réaction croisée entre
les auto-antigènes ARNt synthétases et
les auto-antigènes Ro/SSA retrouvé dans
le Gougerot, a été exclu car il n’y a pas de
similitudes reconnues entre ces 2 types
d’antigènes. Notre patiente avait tous les
critères en faveur du syndrome de Gougerot-
sjögren : cliniques (xérophtalmie, xéros-
tomie, test de Shirmer pathologique),
immunologiques (anticorps anti-Ro/
SSA positifs), et histologiques (sialadé-
nite stade IV de Chisholm et Masson),
ce qui a permis de poser le diagnostic.
L’évolution sous traitement est marquée
le plus souvent par une réponse aux
corticoïdes donnés initialement à raison
de 1 mg/kg par 24 heures équivalent
prednisone. La décroissance du traite-
ment est lentement progressive et guidée
par l’état respiratoire (clinique, radio-
graphique et aux explorations fonction-
nelles), et aussi par les manifestations
extra-respiratoires (articulaire et muscu-
laire). Le traitement doit être prolongé,
mais la dose seuil suffisante pour empê-
cher un rebond pulmonaire ou extra-
pulmonaire varie selon les patients [9].
Chez notre patiente, une réapparition des
arthralgies, voire des arthrites a été notée
lors de la dégression de la corticothéra-
pie. Il est souvent nécessaire d’adjoindre
un immunosuppresseur à titre d’épargne
cortisonique. Cependant, la réponse au
traitement par ces immunosuppresseurs
semble hétérogène, car il y a peu de
données sur l’évolution à long terme, et
aucun facteur prédictible d’évolution n’a
été démontré [3,14,15]. Des études plus
larges avec un suivi prolongé sont néces-
saires pour trouver les meilleurs para-
mètres au moment du diagnostic pour
déterminer le pronostic de ces patients.
CONCLUSION
Le syndrome des antisynthétases à
anti-PL12 est une entité rare au sein
des syndromes des antisynthétases. Il
possède des particularités cliniques et
biologiques qui sont nettement moins
connues vu la rareté des données de la
littérature concernant ce syndrome.
Notre cas illustre une myosite de
chevauchement entre syndrome des
antisynthétases à anti-PL12 et syndrome
de Gougerot-Sjögren. L’association à
cette connectivite est décrite dans la
littérature, avec une fréquence qui
n’est pas négligeable. Ce qui sug-
gère que cette association n’est pas
fortuite, mais le mécanisme étiopatho-
génique reste à élucider. L’évolution
est variable sous traitement, et il n’y
a pas de facteurs prédictibles d’ag-
gravation. Un suivi plus prolongé et
le recueil d’autres observations sont
nécessaires pour évaluer plus précisé-
ment le pronostic de ce syndrome.
Déclaration d’intérêt
Les auteurs déclarent ne pas avoir de
conflit d’intérêt.
RéféRences
[1] Furlan A, Botsios C, Ruatti A, Todesco S, Punzi L. Syndrome des antisynthétases avec polyarthrite réfractaire
et èvre, traité avec succès avec l’anakinra, antagoniste du récepteur de l’IL1. Revue du Rhumatisme
2008;75:504-10.
[2] B. Hervier B, M. Hamidoub M, Y. Uzunhanc Y et al. Comparaison du syndrome des antisynthétases avec anti-
corps antiPL12 (26 patients) avec le syndrome des antisynthétases avec anticorps antiJO1 (50 patients).
Rev Med Interne 2010;31S:S35-S83.
[3] Hervier B, Wallaert B, Hachulla E et al. Clinical manifestations of anti-synthetase syndrome positive for anti-
alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford)
2010;49:972-6.
[4] Mimori T, Imura Y, Nakashima R, Yoshifuji H. Autoantibodies in idiopathic inammatory myopathy: an
update on clinical and pathophysiological signicance. Curr Opin Rheumatol 2007;19:523-9.
[5] Imbert-Masseau A, Hamidou M, Agard Ch, Grolleau JY, Chérin P. Le syndrome des antisynthétases. Revue du
Rhumatisme 2003;70:363-70.
[6] Kalluri M, Sahn SA, Oddis CV et al. Clinical prole of anti-PL-12 autoantibody: cohort study and review of the
literature. Chest 2009;135:1550-6.
[7] Hervier B, Couret B, Adouec D et al. Phénotype clinicobiologique du syndrome des antisynthétases anti-PL12
positif. Rev Med Interne 2008;29S:S294-S336.
[8] Sordet C, Goetz J, Sibilia J. Intérêt diagnostique et nosologique des autoanticorps dans les myopathies
inammatoires. Revue du Rhumatisme 2006;73:1301-10.
[9]. D.Valeyre. Syndrome des antisynthétases. La revue de médecine interne 25 (2004) S17–S18.
[10] Takahashi T, Wada I, Ohtsuka Y, Munakata M, Homma Y, Kuroki Y. Autoantibody to alanyl-tRNA synthetase
in patients with idiopathic pulmonary brosis. Respirology 2007;12:642-53.
[11] Handa T, Nagai S, Kawabata D et al. Long-term clinical course of a patient with anti PL-12 antibody accom-
panied by interstitial pneumonia and severe pulmonary hypertension. Intern Med 2005;44:319-25.
[12] Hirakata M. Autoantibodies to aminoacyl-tRNA synthetases. Intern Med 2005;44:527-8.
[13] Limaye VS, Cassidy J, Scott G, Roberts-Thomson PJ, Gillis D. Anti-Ro52 antibodies, antisynthetase antibodies,
and antisynthetase syndrome. Clin Rheumatol 2008;27:521-3.
[14] Yoshifuji H, Fujii T, Kobayashi S et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course predic-
tion of interstitial lung disease complicated with idiopathic inammatory myopathies. Autoimmunity
2006;39:233-41.
[15] Troyanov Y, Targo IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classication of idiopathic
inammatory myopathies based on overlap syndrome features and anti-bodies: analysis of 100 French
Canadien patients. Medicine (Baltimore) 2005;84:231-49.
Chevauchement entre syndrome des antisynthétases à anti-PL12 et syndrome de Gougerot-Sjögren
47
1
/
3
100%