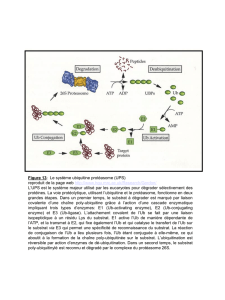
4. Essais enzymatiques

154
4. Essais enzymatiques
Principe général d’un essai enzymatique:
analyte produit(s)
Des matrices biologiques peuvent contenir des espèces qui
interfèrent avec une mesure directe de la concentration d’analyte.
Une méthode enzymatique permettrait de suivre une diminution
(substrat) ou augmentation (produit) du signal (absorbance,
fluorescence, courant) due à la conversion sélective de l’analyte
en produit(s), évitant ainsi une interférence analytique.
enzyme
(substrat)
155
Exemple 4.1: consommation du substrat
Pour quantifier l’acide urique, on mesure la diminution de A293nm.
Exemple 4.2: accumulation du produit
Pour quantifier la coenzyme A, on mesure l’augmentation de A232nm.
HN
N
HN
H
NH
O
OON
H
NH
O
O
HNH2NC
O
+ O2 + 2H2O+ CO2 + H2O2
oxydase
d'urate
ε293nm=1.22x104M-1 cm-1
CoA-SH + CH3CO-OPO3H CoA-S-COCH3+ H3PO4
ε232nm=4.5x103M-1 cm-1
phosphotransacétylase

156
Pour certaines réactions enzymatiques, le changement de signal due à la
consommation du substrat ou l’accumulation du produit peut être utilisé.
Le choix d’espèce dépend des propriétés du substrat et du produit et de la
technique de mesure employée; une comparaison des sensibilités et limites
de détection s’impose.
Par exemple, si le substrat et produit sont tous les deux fluorescents et
possèdent des absorptivités molaires et des rendements quantiques
comparables, mais des longueurs d’onde d’excitation et d’émission
différentes, la quantification du produit est préférable puisqu’une petite
augmentation de la fluorescence est facilement mesurable sur un signal de
fond quasi-nul.
La décision devient plus compliquée dans le cas de la spectroscopie
d’absorption si le substrat et le produit possèdent des absorptivités molaires
égales. Puisque des spectromètres d’absorption mesurent la quantité de
lumière transmisse par un échantillon, le plus grand signal (avec une
précision plus petite) est obtenu à des concentrations faibles d’analyte.
Même si l’on choisit normalement de mesurer l’accumulation du produit en
spectrophotométrie d’absorption, il est important de comparer les courbes
de calibration pour la consommation du substrat et l’accumulation du
produit afin d’identifier la méthode optimale.
157
4.1 Mesures directes et couplées
Certaines réactions enzymatiques peuvent être suivies directement
(consommation du substrat ou accumulation du produit) avec une
précision adéquate pour un essai enzymatique direct.
Par contre, plusieurs enzymes catalysent des réactions impliquant
des espèces qui ne sont pas facilement détectables.
Dans ces situations, le produit est converti en une espèce
détectable dans une réaction subséquente («réaction couplée»
ou «réaction indicatrice»):
Analyte Produit primaire Produit détecté
La réaction indicatrice peut être de nature chimique ou enzymatique;
le critère principal étant que la conversion du produit primaire en
produit détecté doit être rapide et quantitative.
Eprim Eind

158
Exemple 4.3:
N
NN
N
NH2
Ribose
HN
NN
N
Ribose
O
AMP
O
O
OH
OH
P
O
O
N
O
O
P
NH3 + 2
λmax=546 nm
+ NH3
adénosine
désaminase
159
Enzymes indicatrices
Les déshydrogénases:
Substrat + NAD(P)+Substrat + NAD(P)H + H+
(forme réduite) (forme oxydée)
Les déshydrogénases sont utilisées lorsque la réaction enzymatique
primaire produit une espèce qui peut servir comme substrat pour
une réaction de déshydrogénase particulière. Cette espèce est
convertie en sa forme oxydée
dans la réaction indicatrice,
où le NADH ou NADPH formé
peut être détecté a 340 nm
(ε340nm=6.2x103M-1 cm-1).
déshydrogénase
(D’après Mikkelsen & Cortòn,
Bioanalytical Chemistry, 2004)

160
Les peroxydases:
Colorant + H2O2Colorant + H2O
(forme réduite) (forme oxydée)
Des réactions indicatrices à base de peroxydase sont utiles pour
suivre une réaction primaire produisant du peroxyde d’hydrogène.
Malgré la grande spécificité des peroxydases pour le H2O2, elles
réagissent avec une variété d’espèces chromogènes qui sont
incolores dans leur forme réduite, mais ont une forte absorption
dans la forme oxydée (ex. 2,4-dichlorophénol, O-dianisidine (3,3’-
diméthoxybenzidine), benzidine (diamino-4,4’-biphényl), vert
malachite).
peroxydase
161
Deux essais enzymatiques commerciaux pour le glucose dans le sérum
sanguin utilisent la déshydrogénase et la peroxydase:
β-D-glucose + ATP glucose-6-phosphate (G6P) + ADP
G6P + NADP+acide 6-phosphoglycérique + NADPH + H+
O
OH OH
OH
OH
CH2OH O
OH OH
OH
CH2OH
O
+ O2+ H2O2
o
x
y
da
s
e
de glucose
β-D-glucose acide gluconique
H2O2 + O-dianisidine 2,2'-diméthoxybiphénylquinonediimine + 2H2O
ε450nm=8.6x103M-1cm-1
peroxydase
hexokinase
désydrogénase
ε340nm= 6.2x103M-1 cm-1

162
Seules les concentrations de l’analyte et du produit
primaire devraient limiter les vitesses des réactions
couplées. Un excès de tous les autres réactifs est
utilisé.
Pour une conversion linéaire du produit primaire en
produit détecté, des faibles concentrations du produit
primaire doivent être produites (i.e. région linéaire de
la courbe de cinétique Vo-[S]; [S]<0.1Km)
Dans des conditions optimales, la vitesse de la
réaction indicatrice (Veff)ind = vitesse de formation du
produit primaire (Vmax)prim.
Puisque Km,P1 est une caractéristique de Eind
et [P1] est dictée par [analyte], les seules variables
qui peuvent être contrôlées expérimentalement sont
(Vmax)ind ( α [Eind]) et la concentration du co-substrat,
[S2]. Un grand excès (100 fois ou plus) de Eind est
employé, ainsi qu’une concentration saturante du co-
substrat (ex. O-dianisidine ou NADP+).
Analyte
Produit primaire (P1)
+ co-substrat (S2)
Produit détecté
Eprimaire
Eindicatrice
(Vmax)ind
(Vmax)prim
])2S/[(])1P/[1( )(
)()( S2m,P1m,
indmax
indeffprimmax KK V
VV ++
==
163
4.2 Classification des méthodes enzymatiques
Si [S] < 0.1Km, Voα[S] condition pour la quantification du substrat
Si [S] > 10Km, Voα[E] condition pour la quantification d’enzyme
Types d’essais enzymatiques: - essai cinétique
- essai à temps fixe
- essai à changement fixe
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%