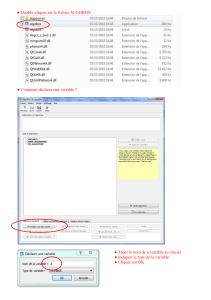
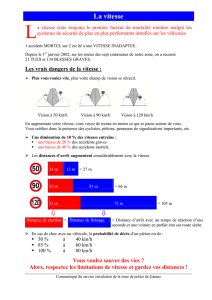
UN NOUVEL ALGORITHME BIOINFORMATIQUE POUR L

UN NOUVEL ALGORITHME
BIOINFORMATIQUE POUR
L'INFÉRENCE DE RÉSEAUX
D'HYBRIDATION EXPLICITES
Matthieu Willems
10 février 2015

PLAN
1. Rappels sur l'inférence phylogénétique.
2. Évolution réticulée.
3. Réseaux phylogénétiques.
4. Un nouvel algorithme.
5. Applications.

1. Inférence phylogénétique
Objectif
Objectif : reconstruire l'histoire évolutive d'un ensemble
d'espèce à partir de données moléculaires

Entrées des algorithmes
phylogénétiques
• Méthodes basées sur les caractères
(séquences moléculaires) :
E1 : ATCGGATCGTATTCGA
E2 : ATGCCTAGGATATGGT
E3 : TTGGGAACGCATCCTA
• Matrices de distances :
E1 E2 E3
E1 0 11 6
E2 11 0 12
E3 6 12 0

Principales méthodes
1. Méthodes de distances (NJ, UPGMA).
2. Maximum de parcimonie.
3. Maximum de vraisemblance.
4. Méthodes bayésiennes.
5. Peu de nouveaux développements
depuis 15 ans.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
1
/
31
100%