Télécharger le texte intégral - Thèses

ÉCOLE NATIONALE VETERINAIRE D’ALFORT
Année
CONTRIBUTION A L’ETUDE DE LA SCLEROSE EN
PLAQUES : UN MODELE MURIN TRANSGENIQUE
THESE
Pour le
DOCTORAT VETERINAIRE
Présentée et soutenue publiquement devant
LA FACULTE DE MEDECINE DE CRETEIL
le……………
par
CABARROCAS Julie, Marie, Cécile
Né (e) le 04 Avril 1975 à Evry (Essonne)
JURY
Président : M.
Professeur à la Faculté de Médecine de CRETEIL
Membres
Directeur : Mme QUINTIN-COLONNA Françoise
Professeur de Pathologie Générale, Microbiologie, Immunologie
Assesseur : M PARODI André-Laurent
Directeur honoraire

LISTE DES MEMBRES DU CORPS ENSEIGNANT
Directeur : M. le Professeur COTARD Jean-Pierre
Directeurs honoraires : MM. les Professeurs PARODI André-Laurent, PILET Charles
Professeurs honoraires: MM. BORDET Roger,BUSSIERAS Jean,LE BARS Henri, MILHAUD Guy,ROZIER Jacques,THERET Marcel,VUILLAUME Robert
DEPARTEMENT DES SCIENCES BIOLOGIQUES ET PHARMACEUTIQUES (DSBP)
Chef du département : M. BOULOUIS Henri-Jean, Professeur - Adjoint : M. DEGUEURCE Christophe, Professeur
-UNITE D’ANATOMIE DES ANIMAUX DOMESTIQUES
Mme CREVIER-DENOIX Nathalie, Professeur*
M. DEGUEURCE Christophe, Professeur
Mlle ROBERT Céline, Maître de conférences
M. CHATEAU Henri, AERC
-UNITE DE PATHOLOGIE GENERALE , MICROBIOLOGIE,
IMMUNOLOGIE
Mme QUINTIN-COLONNA Françoise, Professeur*
M. BOULOUIS Henri-Jean, Professeur
Mme VIALE Anne-Claire, Maître de conférences
-UNITE DE PHYSIOLOGIE ET THERAPEUTIQUE
M. BRUGERE Henri, Professeur *
Mme COMBRISSON Hélène, Professeur
M. TIRET Laurent, Maître de conférences
-UNITE DE PHARMACIE ET TOXICOLOGIE
Mme ENRIQUEZ Brigitte, Professeur *
Mme HUYNH-DELERME, Maître de conférences contractuel
M. TISSIER Renaud, Maître de conférences
- UNITE D’HISTOLOGIE , ANATOMIE PATHOLOGIQUE
M. CRESPEAU François, Professeur *
M. FONTAINE Jean-Jacques, Professeur
Mme BERNEX Florence, Maître de conférences
Mme CORDONNIER-LEFORT Nathalie, Maître de conférences
-UNITE DE BIOCHIMIE
M. BELLIER Sylvain, Maître de conférences*
M. MICHAUX Jean-Michel, Maître de conférences
- UNITE DE VIROLOGIE
M. ELOIT Marc, Professeur *
Mme ALCON Sophie, Maître de conférences contractuel
-DISCIPLINE : PHYSIQUE ET CHIMIE BIOLOGIQUES ET
MEDICALES
M. MOUTHON Gilbert, Professeur
-DISCIPLINE : BIOLOGIE MOLECULAIRE
Melle ABITBOL Marie, Maître de conférences contractuel
-DISCIPLINE : ETHOLOGIE
M. DEPUTTE Bertrand, Professeur
DEPARTEMENT D’ELEVAGE ET DE PATHOLOGIE DES EQUIDES ET DES CARNIVORES (DEPEC)
Chef du département : M. FAYOLLE Pascal, Professeur - Adjointe : Mme BEGON Dominique , Professeur
-UNITE DE MEDECINE
M. POUCHELON Jean-Louis, Professeur*
M. CLERC Bernard, Professeur
Mme CHETBOUL Valérie, Professeur
M. MORAILLON Robert, Professeur
M. BLOT Stéphane, Maître de conférences
M. ROSENBERG Charles, Maître de conférences contractuel
Melle MAUREY Christelle, Maître de conférences contractuel
- UNITE DE CLINIQUE EQUINE
M. DENOIX Jean-Marie, Professeur *
M. TNIBAR Mohamed, Maître de conférences contractuel
M. AUDIGIE Fabrice, Maître de conférences
Mme DESJARDINS-PESSON Isabelle, Maître de confér..contractuel
-UNITE DE REPRODUCTION ANIMALE
M. MIALOT Jean-Paul, Professeur * (rattaché au DPASP)
M. NUDELMANN Nicolas, Maître de conférences
Mme CHASTANT-MAILLARD Sylvie, Maître de conférences
(rattachée au DPASP )
M. FONTBONNE Alain, Maître de conférences
M. REMY Dominique, Maître de conférences (rattaché au DPASP)
Melle CONSTANT Fabienne, AERC (rattachée au DPASP)
- UNITE DE PATHOLOGIE CHIRURGICALE
M. FAYOLLE Pascal, Professeur *
M. MAILHAC Jean-Marie, Maître de conférences
M. MOISSONNIER Pierre, Professeur
Mme VIATEAU-DUVAL Véronique, Maître de conférences
M. DESBOIS Christophe, Maître de conférences
Mlle RAVARY Bérangère, AERC (rattachée au DPASP)
M. ZILBERSTEIN Luca, Maître de Conférences contractuel
M. HIDALGO Antoine, Maître de Conférences contractuel
- UNITE DE RADIOLOGIE
Mme BEGON Dominique, Professeur*
M. RUEL Yannick, AERC
- UNITE DE PARASITOLOGIE ET MALADIES PARASITAIRES
M. CHERMETTE René, Professeur *
M. POLACK Bruno, Maître de conférences
M. GUILLOT Jacques, Professeur
Melle MARIGNAC Geneviève, Maître de conférences contractuel
M. PARAGON Bernard, Professeur (rattaché au DEPEC)
M. GRANDJEAN Dominique, Professeur (rattaché au DEPEC)
DEPARTEMENT DES PRODUCTIONS ANIMALES ET DE LA SANTE PUBLIQUE (DPASP)
Chef du département : M. CERF Olivier, Professeur - Adjoint : M. BOSSE Philippe, Professeur
-UNITE DES MALADIES CONTAGIEUSES
M. TOMA Bernard, Professeur
M. BENET Jean-Jacques, Professeur*
Mme HADDAD H0ANG XUAN Nadia, Maître de confér.contractuel
M. SANAA Moez, Maître de conférences
-UNITE D’HYGIENE ET INDUSTRIE DES ALIMENTS
D’ORIGINE ANIMALE
M. BOLNOT François, Maître de conférences *
M. CARLIER Vincent, Professeur
M. CERF Olivier, Professeur
Mme COLMIN Catherine, Maître de conférences
M. AUGUSTIN Jean-Christophe, Maître de conférences
- UNITE DE ZOOTECHNIE, ECONOMIE RURALE
M. BOSSE Philippe, Professeur
M. COURREAU Jean-François, Professeur*
Mme GRIMARD-BALLIF Bénédicte, Maître de conférences
Mme LEROY Isabelle, Maître de conférences
M. ARNE Pascal, Maître de conférences
M. PONTER Andrew, Maître de conférences
- UNITE DE PATHOLOGIE MEDICALE DU BETAIL ET DES
ANIMAUX DE BASSE-COUR
Mme BRUGERE-PICOUX Jeanne, Professeur
M.MAILLARD Renaud, Maître de conférences associé
M. MILLEMANN Yves, Maître de conférences*
M. ADJOU Karim, Maître de conférences
Ingénieurs Professeurs agrégés certifiés (IPAC) : * Responsable de l’Unité
Mme CONAN Muriel, Professeur d’Anglais
Mme CALAGUE, Professeur d’Education Physique AERC : Assistant d’Enseignement et de Recherche Contractuel

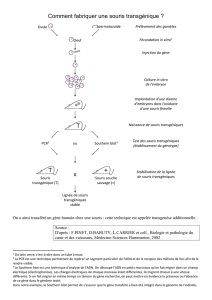
Contribution à l’étude de la sclérose en plaques : un modèle
murin transgénique
NOM et Prénom : CABARROCAS Julie, Marie, Cécile
La physiopathologie de la sclérose en plaques, une maladie inflammatoire et démyélinisante
du système nerveux central (SNC), est encore mal connue mais plusieurs arguments suggèrent
qu’une réponse auto-immune ciblant un (des) antigène(s) du SNC serait impliquée dans le
développement des lésions. Nous avons évalué le potentiel pathogène de lymphocytes T (LT)
CD8+ spécifiques d’un antigène du système nerveux à l’aide de deux lignées de souris
transgéniques. Dans un modèle induit d’affection auto-immune, nous avons mis en évidence
le potentiel encéphalitogène de LT CD8+ et leur aptitude à détruire les cellules exprimant
l’antigène cible dans le SNC avec une extrême sélectivité. De plus, l’étude d’un modèle
spontané de destruction auto-immune des cellules gliales du système nerveux entérique a
révélé que ces cellules pourraient jouer un rôle central dans le maintien de l’intégrité de la
paroi intestinale.
Mots clés : sclérose en plaques, lymphocyte T, système nerveux, maladie auto-immune,
expérimentation animale, modèle animal, animaux transgéniques, rongeur souris.
Jury :
Président : Pr.
Directeur : Pr. QUINTIN-COLONNA Françoise
Assesseur : Pr. PARODI André-Laurent
Invité :
Adresse de l’auteur :
40 rue Victor Hugo
94700 MAISONS-ALFORT
FRANCE

Contribution to the study of the pathophysiology of multiple
sclerosis: a transgenic mouse model
SURNAME : CABARROCAS Julie, Marie, Cécile
The pathophysiology of multiple sclerosis, a chronic inflammatory and demyelinating disease
of the central nervous system (CNS), is not fully understood, yet some evidence indicate that
an auto-immune process targeting CNS autoantigen(s) might be involved in the development
of the lesions. We have investigated the pathogenic potential of CD8+ T lymphocytes specific
for an autoantigen expressed in the nervous system using two transgenic mouse lines. An
induced model of auto-immune disease allowed us to demonstrate the encephalitogenic
potential of CD8+ T cells and their ability to lyse the cells that express the target antigen in
the CNS with exquisite selectivity. Moreover, the study of a spontaneous model of auto-
immune destruction of enteric glial cells revealed that these cells could play a central role in
the maintenance of the integrity of the gut wall.
Keywords: multiple sclerosis, T cell, nervous system, auto-immune disease, animal
experimentation, animal model, transgenic animal, rodent mouse.
Jury :
Président : Pr.
Director : Pr. QUINTIN-COLONNA Françoise
Assesor : Pr. PARODI André-Laurent
Guest : M.
Author’s address:
40 rue Victor Hugo
94700 MAISONS-ALFORT
FRANCE

AVANT-PROPOS
Les travaux décrits ici ont été réalisés dans le cadre de mon stage de DEA puis
de ma thèse de Sciences, sous la direction du Pr. R. Liblau, au sein des unités
Inserm U546 (CHU Pitié-Salpêtrière, Paris; Unité dirigée par le Dr A. Baron van
Evercooren), puis U563 (CHR Purpan - CPTP, Toulouse ; Unité dirigée par le
Pr. G. Delsol).
Les coupes histologiques, les colorations et les marquages en immunohisto-
chimie et l’analyse histo-pathologique des tissus ont été réalisés dans le cadre
d’une collaboration dans le laboratoire du Pr. H. Lassmann, au Brain Research
Institute de Vienne (Autriche).
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
1
/
52
100%