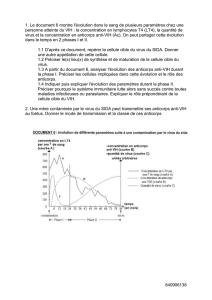
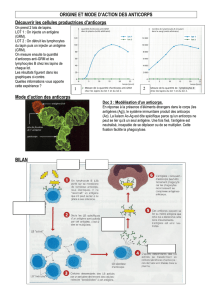
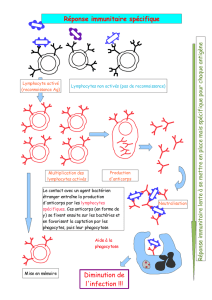
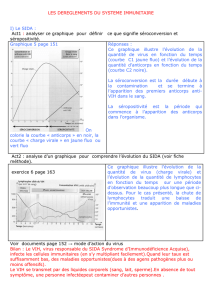
Quand et pourquoi rechercher des anticorps anti

Dans le syndrome de l’homme raide,
les anticorps anti-GAD sont présents
chez plus de 90 % des patients présen-
tant une forme généralisée dite “forme
classique” (5). Ils sont moins fré-
quents dans les autres formes de la
maladie, puisque la fréquence n’est
estimée qu’à 15 % dans les formes
limitées aux membres (6) et seuls
quelques rares cas isolés ont été rap-
portés dans les formes avec encépha-
lite ou avec myoclonus. Les anticorps
anti-GAD sont associés à des formes
idiopathiques du syndrome de
l’homme raide. Dans les formes para-
néoplasiques, les patients présentent
des anticorps anti-amphiphysine (7).
La présence d’anticorps anti-GAD est
exceptionnelle dans ces formes para-
néoplasiques (8). Les anticorps anti-
GAD, retrouvés chez les patients ayant
un syndrome de l’homme raide, pré-
sentent plusieurs caractéristiques qui
les distinguent de ceux qui sont obser-
vés chez les patients diabétiques. Tout
d’abord, le taux d’anticorps est très
élevé, bien supérieur à celui des diabé-
tiques. L’épitope reconnu est différent.
Il est linéaire chez les patients avec
syndrome de l’homme raide et confor-
mationnel chez les diabétiques. De ce
fait, les anticorps anti-GAD des
patients avec syndrome de l’homme
raide peuvent être mis en évidence par
western-blot. Enfin, le mode d’activa-
tion du système immunitaire semble
différent dans les deux groupes. Chez
les diabétiques, on observe une activa-
tion majoritaire de la voie Th1, avec
augmentation de l’interleukine 1 et de
l’interféron gamma, tandis que chez
les patients avec un syndrome de
l’homme raide, c’est la voie Th2, avec
augmentation des interleukines 6 et 4,
qui est activée. De nombreux éléments
cliniques et expérimentaux sont en
faveur d’une atteinte élective du sys-
tème GABA-ergique des interneu-
rones de la moelle épinière comme
étant à l’origine du syndrome de
l’homme raide. Une étude récente,
effectuée in vitro, a montré que les
anticorps anti-GAD des patients avec
syndrome de l’homme raide pouvaient
réduire l’activité enzymatique de la
GAD et donc la synthèse de GABA,
contrairement aux anti-GAD de
patients diabétiques qui n’ont aucun
effet (9). Ce résultat suggère que les
anticorps anti-GAD pourraient avoir
un effet pathogénique direct sur la
moelle épinière des patients, mais
cette hypothèse reste à démontrer in
vivo. L’implication du système immu-
nitaire dans la survenue des troubles
semble néanmoins très probable chez
les patients avec anticorps anti-GAD.
En effet, la fréquence des maladies
auto-immunes (dysthyroïdies, ané-
mies auto-immunes, diabète, etc.) est
particulièrement élevée dans ce
groupe (60 %), alors qu’elle n’est que
de 6 % chez les patients avec syn-
drome de l’homme raide, mais sans
anti-GAD. Il est donc possible qu’il
puisse exister plusieurs mécanismes
pouvant conduire à la survenue d’un
syndrome de l’homme raide, l’un
auto-immun et l’autre non. Dans ce
cas, la mise en évidence d’anticorps
anti-GAD serait particulièrement
importante, car elle permettrait de
sélectionner les patients susceptibles
de répondre à un traitement immuno-
186
Examen rare complémentaire
Examen rare complémentaire
Quand et pourquoi rechercher
des anticorps anti-GAD en
neurologie ?
J. Honnorat*
Jérôme Honnorat neurologue, PU-PH de
Neurologie et travaille dans le service
de neurologie du Professeur Trouillas à
Lyon. Il a beaucoup travaillé sur les syn-
dromes paranéoplasiques et a récem-
ment publié sur les anticorps anti-GAD
(Arch Neurol 2001)
a glutamate décarboxylase
(GAD) est une enzyme
majeure du système nerveux
qui catalyse la conversion du gluta-
mate en GABA. Cette enzyme est
également exprimée dans les cellules
bêta du pancréas et a été identifiée
comme un antigène majeur et essen-
tiel au développement du diabète
insulino-dépendant (DID) (1). Les
anticorps anti-GAD sont présents
chez plus de 80 % des patients pré-
sentant un DID et peuvent être
détectés plusieurs années avant le
début clinique de la maladie, ce qui
permet de les utiliser comme mar-
queur pronostique (2). Chez les
patients non diabétiques, il est
exceptionnel de détecter ces anti-
corps. Néanmoins, des taux élevés
ont également été retrouvés chez
quelques rares patients présentant
des troubles neurologiques. Ces anti-
corps anti-GAD pourraient jouer un
rôle direct et majeur dans la survenue
de ces syndromes. Actuellement,
deux syndromes neurologiques asso-
ciés aux anticorps anti-GAD sont clai-
rement identifiés : le syndrome de
l’homme raide (3), l’ataxie cérébel-
leuse d’apparence dégénérative (4). Il
est également possible qu’il existe
d’autres syndromes neurologiques
associés à ces anticorps, non encore
identifiés.
L

187
Examen rare complémentaire
Examen rare complémentaire
Act. Méd. Int. - Neurologie (3) n° 8, octobre 2002
suppresseur. Cette hypothèse reste à
confirmer.
Les patients présentant une ataxie
cérébelleuse et des anticorps anti-
GAD sont rares. Moins d’une ving-
taine de cas ont été rapportés dans la
littérature, mais la présentation cli-
nique est très stéréotypée, suggérant
un syndrome particulier (4). Il s’agit
majoritairement de femmes (90 %)
avec un âge moyen de 55 ans au début
de la symptomatologie cérébelleuse.
Une histoire familiale de maladie
auto-immune telle que DID ou mala-
dies thyroïdiennes est fréquente (plus
de 50 % des cas). Un diabète insulino-
requérant de début tardif est présent
chez plus de 71 % des patients, et
d’autres maladies auto-immunes,
comme des thyroïdites (hyper- ou
hypothyroïdie), des anémies hémoly-
tiques, des myasthénies, des thy-
momes malins, des psoriasis ou des
maladies cœliaques, ont également été
rapportées. Le syndrome cérébelleux,
essentiellement statique, s’installe en
général progressivement, suggérant
une atteinte dégénérative. Un nystag-
mus et une dysarthrie sont fréquents et
il a également été rapporté une rigidité
d’un membre, suggérant un syndrome
de l’homme raide focalisé, une neuro-
pathie périphérique ou une myasthé-
nie. Le liquide céphalorachidien est en
principe normal pour ce qui est du
taux de protéine ou du nombre de cel-
lules, mais il est important de recher-
cher des bandes oligo-clonales, obser-
vées de façon isolée dans 80 % des
cas. Le scanner et l’IRM encéphalique
montrent généralement une atrophie
cérébelleuse pure et diffuse chez plus
de 50 % des patients. Il n’a jamais été
observé d’atrophie du tronc cérébral.
L’ e xistence de bandes oligo-clonales
dans le liquide céphalorachidien de
ces patients, associée à une synthèse
intrathécale d’anticorps anti-GAD,
ainsi qu’à la présence anormalement
élevée d’anticorps spécifiques d’or-
ganes et de maladies auto-immunes,
distingue clairement ces patients des
autres qui présentent une ataxie céré-
belleuse sporadique, et suggère forte-
ment l’implication du système immu-
nitaire dans la survenue de l’ataxie
cérébelleuse. De plus, le fait que cer-
tains patients ont un syndrome de
l’homme raide focalisé, associé à
l’ataxie, suggère que ces deux patho-
logies ont des mécanismes physiopa-
thologiques communs. Comme dans le
syndrome de l’homme raide, le taux
d’anticorps anti-GAD est particulière-
ment élevé et l’épitope reconnu est
différent de celui des patients diabé-
tiques. Les anticorps pourraient égale-
ment jouer un rôle direct dans la sur-
venue de l’ataxie, puisqu’il a été
démontré que le sérum de ces patients
ataxiques pouvait supprimer sélective-
ment la transmission GABA-ergique
sur des coupes de cervelet de rat (10).
De plus, ces patients seraient sensibles
à un traitement par veinoglobulines
(11).
Des taux élevés d’anticorps anti-GAD
ont également été rapportés chez des
patients présentant une épilepsie
réfractaire (12) ou des rétinopathies
(13), mais ces observations sont trop
rares, à l’heure actuelle, pour pouvoir
affirmer qu’il ne s’agit pas d’associa-
tions liées au hasard.
Conclusion
L’association d’un trouble neurolo-
gique et d’anticorps anti-GAD est
rare. Leur recherche est néanmoins
indispensable chez des patients pré-
sentant un syndrome de l’homme
raide ou une ataxie cérébelleuse spora-
dique, surtout s’il existe des antécé-
dents familiaux ou personnels de
maladies auto-immunes. Leur pré-
sence est un argument important pour
évoquer une origine auto-immune à la
survenue des troubles et proposer un
traitement immuno-modulateur.
Néanmoins, l’interprétation du résul-
tat du dosage devra être rigoureuse.
Seuls les taux élevés pourront être
retenus, les taux faibles pouvant être
liés à l’existence d’un diabète latent,
très fréquent dans la population géné-
rale.
Références
1. Yoon JW, Yoon CS, Lim HW et al.
Control of autoimmune diabetes in NOD
mice by GAD expression or suppression in
beta cells. Science 1999 ; 284 : 1183-7.
2. Baekkeskov S, Aanstoot HJ, Christgau S
et al. Identification of the 64K autoantigen
in insulin-dependent diabetes as the
GABA-synthesizing enzyme glutamic acid
decarboxylase. Nature 1990 ; 347 : 151-6.
3. Solimena M, Folli F, Aparisi R et al.
Autoantibodies to GABA-ergic neurons
and pancreatic beta cells in stiff-man syn-
drome. N Engl J Med 1990 ; 322 : 1555-60.
4. Honnorat J, Saiz A, Giometto B et al.
Cerebellar ataxia with anti-glutamic acid
decarboxylase antibodies : clinical and
immunological data of a series of 14
patients. Arch Neurol 2001 ; 58 : 225-30.
5. Brown P, Marsden CD.The stiff man and
stiff man plus syndrome. J Neurol 1999 ;
246 : 648-52.
6. Barker RA, Revesz T, Thom M et al.
Review of 23 patients affected by stiff-man
syndrome : clinical subdivision into stiff
trunk (man) syndrome, stiff-limb syn-
drome, and progressive encephalomyelitis
with rigidity. J Neurol Neurosurg
Psychiatry 1998 ; 65 : 633-40.
7. Folli F, Solimena M, Cofiell R et al.
Autoantibodies to a 128-kd protein in three
women with the stiff-man syndrome and
breast cancer. N Engl J Med 1993 ; 328 :
546-51.
8. Silverman IE. Paraneoplastic stiff limb
syndrome. J Neurol Neurosurg Psychiatry
1999 ; 67 : 126-7.
9. Dinkel K, Meinck HM, Jury KM et al.
Inhibition of aminobutyric acid synthesis
by glutamic acid decarboxylase autoanti-
bodies in stiff-man syndrome. Ann Neurol
1998 ; 44 : 194-201.
10. Ishida K, Mitoma H, Song SY et al.
Selective suppression of cerebellar GABA-
ergic transmission by an autoantibody to
glutamic acid decarboxylase. Ann Neurol
1999 ; 46 : 263-7.
11. Abele M, Weller M, Mescheriakov S et
al. Cerebellar ataxia with glutamic acid
decarboxylase autoantibodies. Neurology
1999 ; 52 : 857-9.
12. Peltola J, Kulmala P, Isojarvi J et al.
Autoantibodies to glutamic acid decar-
boxylase in patients with therapy-resistant
epilepsy. Neurology 2000 ; 55 : 46-50 .
13. Steffen H, Menger N, Richter W et al.
Immune-mediated retinopathy in a patient
with stiff-man syndrome. Graefe’s Arch
Clin Exp Ophtalmol 1999 ; 237 : 212-9.
1
/
2
100%