Présentation PowerPoint

Page 20 / 30
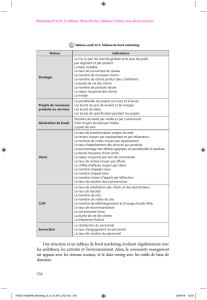
Tous les paramètres mesurés lors de l’étape précédente serviront à optimiser le potentiel de développement
de la molécule. La connaissance des relations structure-activité permettra aux chimistes et chémo-
informaticiens d’établir un plan d’optimisation chimique pour ces différents paramètres.
Le cahier des charges décrivant les caractéristiques requises doit obligatoirement être établi (compound profil
ou compound Report Card). Il devra décrire les objectifs chiffrés attendus et relatifs au projet, à la pathologie
ciblée, à la voie d’administration envisagée, à l’index thérapeutique acceptable, à l’organe ciblé, à la toxicité
attendue, à l’activité minimale et au profil pharmacocinétique attendu.
OBJECTIFS
Le but est d’optimiser l’activité pharmacologique (ex : affinité, sélectivité ou polypharmacologie) et la capacité
des leads à devenir candidat médicament (en sélectionnant au besoin une molécule alternative, « back-up »)
selon le cahier des charges établi. A ce stade, les chercheurs préciseront encore la définition les niveaux
d’affinité, d'efficacité, de sélectivité et de pharmacocinétique. Les multiples analogues préparés renforcement
les relations structure-activité. Cette étape peut également conduire à l’identification de paires de molécules
de structures proches mais d’activité très différente, « activity cliff ». Ces molécules sont précieuses pour
guider ensuite les étapes de drug design et définir les régions moléculaires à modifier. Ce processus est
susceptible de conduire à d’autres générations d’analogues aux propriétés améliorées ; les meilleures d’entre
eux seront à nouveau testés sur des modèles animaux ou systèmes « in vitro- in cellulo », jugés pertinents
pour la pathologie étudiée.
NOMBRE DE MOLÉCULES
Une dizaine voire quelques dizaines de molécules par lead selon le temps et les moyens à disposition.
Comme les coûts sont en général significatifs une assistance chémo-informatique peut, avant la synthèse,
s’avérer précieuse afin de prédire certaines propriétés (affinité, sélectivité et ADME-Tox - Absorption,
Distribution, Métabolisme, Excrétion Toxicité) et d’explorer rapidement un grand nombre d’hypothèses pour
concentrer les efforts expérimentaux sur les molécules prometteuses.
CRITÈRES DE SÉLECTION
Les critères de sélection dépendent de la pathologie ciblée et de la durée du traitement (chronique vs aigu),
l’index thérapeutique « tolérable » en dépendra et donc l’optimisation prioritaire des différents paramètres.
Le cahier des charges établi ci-dessous doit être respecté. Les paramètres physico-chimiques et biologiques
(biodisponibilité, toxicité…) sont aussi déterminants que ceux qui régissent l’activité pharmacologique et
constituent des « Go/NoGo ».
1. Etudes biophysiques (RMN, radiocristallographie, résonance plasmonique de surface,…)
2. Etude in silico avec une approche multiparamétrique afin d’optimiser activité et propriétés ADMET
3. Etudes de pharmacocinétique (ADME : absorption, distribution, métabolisme et excrétion)
4. Etude de pré-formulation et formulation (la meilleure formulation sera choisie en fonction de l'étude
pharmacocinétique)
5. Etudes de pharmaco-dynamie (activité)
6. Etudes de pharmaco-toxicologie (genotoxicité, cardiotoxicité, cytotoxicité…)
Il est fortement recommandé d’avoir, à ce stade, breveté la série chimique et son(ses) application(s), en
raison des multiples partenariats qui deviendront nécessaires au cours des étapes suivantes.
Optimisation des leads : du lead au candidat médicament
5
TRL 4

Page 21 / 30
Privé Public
-
AFSSI afssi.fr
-
Atlanbio atlanbio.com (Pays de Loire)
-
Atlanchim pharma atlanchimpharma.com (Pays de Loire)
-
BCI Pharma www.bci-pharma.com (Languedoc Roussillon)
-
Bertin Pharma www.bertinpharma.com (Ile de France)
-
Capeval Pharma capeval-pharma.com (Rhone Alpes)
-
C-Rispharma c-rispharma.com (Bretagne)
-
DiverChim diverchim.com (Ile de France)
-
Drugabilis drugabilis.com (Ile de France)
-
NatXRay natx-ray.com (Rhone Alpes)
-
Provence Technologies provetech.com (PACA)
Plates-formes GIS Ibisa (ibisa.net) :
-TechMedill (Strasbourg)
-CEA/SPI-LEEM (CEA)
-Prim (Lille)
-Primacen (Rouen)
-ImPACcell (Rennes)
-CHEM-Symbiose (Nantes)
-Synbio3 (Montpelier)
Ainsi que :
-SynBioN (Nancy)
-Cobra (Rouen)
En conclusion l’optimisation du lead aboutit à un candidat médicament (et à la molécule « back-up ») ainsi
défini : une molécule bien caractérisée, « drug-like » et représentant le meilleur compromis pour répondre au
profil attendu (cahier des charges).
La nouvelle entité chimique entrera ensuite dans les phases d’évaluations préclinique réglementaire.
En cas d’échec la molécule back-up peut alors être rapidement évaluée.
Optimisation des leads : du lead au candidat médicament
5
•Holdgate G. et al. (2013) Biophysical methods in drug discovery from small molecule to pharmaceutical. Methods
Mol. Biol., 1008:327-355.
•Segall M.D. (2012) Multi-parameter optimization: identifying high quality compounds with a balance of properties.
Curr. Pharm. Des., 18(9):1292-1310.
•Hefti F.F. (2008) Requirements for a lead compound to become a clinical candidate. BMC Neurosciences, 9:S7
•Hopkins A.L. et al. (2014) The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov.,
13(2):105-121
Références
Ressources ADME Tox
-Amasti amatsi.com
-Biogalenys biogalenys.com
-Biotrial biotrial.com (Rennes, Nantes)
-CERB cerb.fr
-CEREP cerep.fr (Poitiers)
-Chelatec chelatec.fr (Pays de Loire)
-CiToxLAB citoxlab.com (Ile de France)
-Elaia pharm lundbeck.com/elaiapharm (PACA)
-Eurofins eurofins.fr (Nantes/ Montpellier)
-Gimopharm gimopharm.com (Ile de France)
-Oroxell oroxcell.com (Ile de France)
-Prosolt porsolt.com (Pays de Loire)
-Rowin roowin.com (Auvergne)
1
/
2
100%