Physiopathologie des maladies auto-immunes

La Revue de médecine interne 31S (2010) S292–S295
Physiopathologie des maladies auto-immunes
Pathogenic mechanisms of autoimmune diseases
B. Bonnotte
Inserm U866, service d’immunologie clinique et de médecine interne, hôpital du Bocage, CHU de Dijon, faculté de médecine,
2, boulevard Maréchal-de-Lattre-de-Tassigny, 21000 Dijon cedex, France
info article
Historique de l’article :
Disponible sur Internet le 29 octobre 2010
Mots clés :
Maladie auto-immune
Physiopathologie
Lymphocyte T
Cellule dendritique
Keywords:
Autoimmune disease
Physiopathology
T lymphocyte
Dendritic cell
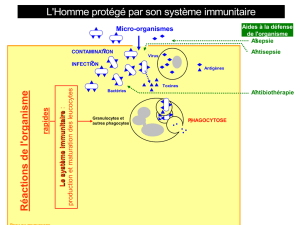
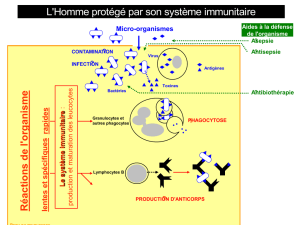
1. La réponse immunitaire
Une maladie auto-immune (MAI) peut se définir par l’activation
du système immunitaire du patient contre ses propres Ag. La
réponse immunitaire spécifique d’Ag, d’auto-Ag en ce qui concerne
les MAI, est orchestrée principalement par deux types de cel-
lules : les cellules présentatrices d’Ag, particulièrement les cellules
dendritiques (DC) et les lymphocytes (Lc). Les DC diffèrent fonc-
tionnellement et phénotypiquement en fonction de leur stade de
maturation. Immatures, les DC phagocytent les Ag. Cependant ces
cellules ne peuvent pas présenter les Ag car elles n’expriment pas
à leur surface en nombre suffisant des molécules de complexe
majeur d’histocompatibilité (CMH), d’adhésion et de costimula-
tion. Lorsque les DC deviennent matures, sous l’influence de divers
signaux «danger », notamment inflammatoires elles perdent leur
capacité de phagocytose mais acquièrent des capacités de cellules
présentatrices. Elles découpent les Ag en petits peptides d’une
dizaine d’acides aminés et les présentent au sein de molécules
de CMH. Parallèlement elles expriment de nombreuses molécules
d’adhésion et de costimulation. Les Lc T qui ont le bon récep-
teur (TCR pour T-cell receptor) vont reconnaître l’Ag présenté par
la DC mature. Cette liaison (TCR-Ag) associée à un second signal
donné par les liaisons entre molécules de costimulation, en parti-
Adresse e-mail : [email protected].
culier CD80 et CD86 exprimées par les DC matures et la molécule
CD28 exprimée par le Lc T va induire l’activation du Lc T. Un Lc T
activé va proliférer et sécréter de nombreuses cytokines qui vont
permettre l’activation de cellules cytotoxiques : macrophages, cel-
lules NK, la différenciation des Lc T CD8+ en Lc T cytotoxiques et
l’activationdesLcB.Le Lc Ba aussi lescapacités de présentationd’Ag
et la coopération entre le Lc T et le Lc B via les molécules de costimu-
lation comme CD28/CD80, CD40L/CD40, OX40L/OX40, ICOS/B7H va
entraîner l’activation et la différentiation du Lc B en plasmocytes
qui vont produire des taux élevés d’anticorps (Fig. 1).
Malgré les connaissances théoriques explicitées dans ce schéma,
la physiopathologie des MAI reste toujours mal connue pour la
majorité d’entre elles. En théorie, un ou plusieurs facteurs rarement
identifiés déclencheraient chez des personnes au terrain génétique
particulier, une prolifération et activation de Lc T et B autoréac-
tifs. De plus, l’association d’atteintes préférentielles de tel ou tel
organe qui nous permet à nous cliniciens de suspecter une patho-
logie précise, n’a pas d’explication rationnelle à ce jour pour la
majorité des maladies systémiques. En effet, en cas d’expression
des auto-Ag dans un seul organe, la maladie sera limitée à cet
organe (exemples : thyroïdite, diabète). À l’inverse si les auto-Ag
sont exprimés de fac¸ on systémique, la MAI sera étiquetée sys-
témique. Mais dans la plupart des maladies systémiques, malgré
une expression ubiquitaire de l’auto-Ag, l’atteinte clinique est très
souvent localisée à certains organes (exemples : Ag ribonucléo-
protéiques Sicca Syndrome A/B (SSA et SSB) dans le syndrome de
0248-8663/$ – see front matter © 2010 Société nationale française de médecine interne (SNFMI). Publié par Elsevier Masson SAS. Tous droits réservés.
doi:10.1016/j.revmed.2010.09.017

B. Bonnotte / La Revue de médecine interne 31S (2010) S292–S295 S293
Fig. 1. La réponse immunitaire : activation des Lc T par les DC et coopération Lc T et Lc B.
Sjögren ou ARNt-synthétases dans la polymyosite). À ce jour, nous
n’avons pas d’explication très précise à ces atteintes d’organe pré-
férentielles hormis les hypothèses de problèmes d’accessibilité à
l’Ag et/ou de migration des cellules du système immunitaire ou
d’expression séquentielle des Ag en fonction de l’âge et/ou du
microenvironnement [1,2].
2. Susceptibilité génétique
Les études génétiques réalisées dans les modèles animaux de
MAI ont montré qu’il existait au moins 25 gènes qui peuvent contri-
buer à une susceptibilité particulière aux MAI. Ces gènes codent
principalement pour les protéines du CMH de classe I et de classe II,
les cytokines, les récepteurs des cytokines, les protéines impliquées
dans la régulation de la réponse immunitaire et dans l’apoptose.
Chez l’homme, la présence de certains allèles du CMH de classe I ou
de classe II est associée à une augmentation du risque de survenue
de certaines pathologies auto-immunes [3]. De rares pathologies
sont fortement liées à un CMH particulier, comme HLA B27et spon-
dylarthrite ankylosante ou HLA DQ2 et DQ8 et maladie cœliaque,
mais dans la plupart des cas l’augmentation du risque liée au CMH
n’est pas très importante, et il paraît probable que la plupart des
MAI sont multigéniques [4,5].
3. Déclenchement de la maladie auto-immune
Même si la prédisposition génétique est un facteur important,
des événements extérieurs sont souvent indispensables pour le
déclenchement d’une MAI. Ainsi, la concordance est relativement
basse entre les jumeaux vrais pour les MAI ; elle n’est que de 33 %
pour le diabète auto-immun et 25 % pour le lupus [6].
Le système immunitaire joue à la fois un rôle dans la destruction
et dans la tolérance des cellules. Ainsi, notre système immunitaire
tolère nos propres cellules. Pour qu’une MAI apparaisse, il faut aussi
une rupture de tolérance immunitaire. Cette tolérance repose sur
un mécanisme central au niveau du thymus (Lc T) et de la moelle
osseuse (Lc B) et sur des mécanismes périphériques.
3.1. Mécanismes de tolérance centrale du thymus
Au sein du thymus les récepteurs des Lc T sont générés grâce
à une recombinaison au hasard de segments géniques. Pour éviter
la survenue de MAI, l’organisme doit éliminer les Lc T fortement
autoréactifs. Au sein du thymus, tous les Ag de l’organisme sont
présentés et tous les Lc T qui reconnaissent fortement un auto-Ag
meurent par apoptose [3,7]. Cette sélection se résume par la phrase
suivante : «L’organisme est tolérant à tout ce que le Lc T voit dans
le thymus ». Le facteur de transcription (qui permet l’activation de
gènes) auto-immune regulator (AIRE) est impliqué dans l’expression
de nombreux gènes codant pour de nombreuses protéines. Aussi
la mutation de AIRE, qui induit une diminution de l’expression de
plus d’une centaine de gènes au sein du thymus, est responsable
du syndrome polyendocrinien de type I (APECED pour autoim-
mune polyendocrinopathy, candidiasis, ectodermal dystrophy) avec
atteinte auto-immune plurisystémique (alopécie, hépatite, anémie,
diabète, vitiligo, atteinte des parathyroïdes et des glandes surré-
nales). Néanmoins, chez la grande majorité des patients atteints de
MAI, aucune anomalie de l’expression de facteurs de transcription
n’a été rapportée à ce jour.
3.2. Rupture de tolérance périphérique
Chez tous les individus il existe dans l’organisme des Lc T recon-
naissant avec une affinité faible des Ag du soi et ces Ag du soi sont
exprimés par les cellules de l’organisme. Néanmoins dans la plupart
des cas, les patients ne développent pas de MAI. Les deux princi-
pales raisons qui expliquent l’absence de MAI sont une présentation
inefficace de l’auto-Ag par les cellules de l’organisme ne permettant
pas l’activation des Lc T autoréactifs et une régulation efficace du
système immunitaire.
3.2.1. Exemples de situations dans lesquelles une meilleure
présentation de l’auto-Ag peut déclencher une maladie
auto-immune
Pour qu’une MAI se déclenche, il faut que des Lc T reconnaissent
un auto-Ag présenté par une cellule présentatrice et rec¸ oivent des

S294 B. Bonnotte / La Revue de médecine interne 31S (2010) S292–S295
signaux d’activation. Ainsi les auto-Ag présentés par des cellules
présentatrices qui n’expriment pas les molécules de costimulation
ne pourront pas déclencher l’activation des Lc T autoréactifs. Pour
déclencher une MAI, un auto-Ag doit donc être efficacement pré-
senté [8] et la meilleure cellule présentatrice d’Ag est la DC.
3.2.1.1. Mimétisme moléculaire. Nos cellules expriment des Ag du
soi et nos Lc T autoréactifs circulent dans l’organisme mais ne
déclenchent aucune MAI car, même s’ils reconnaissent les auto-
Ag, les cellules non immunitaires qui présentent ces auto-Ag sont
incapables de les activer. À l’occasion d’une infection par un micro-
organisme qui exprime des Ag communs avec les auto-Ag du
patient, l’organisme va déclencher une réponse immunitaire qui
va détruire à la fois cet agent infectieux mais aussi ses propres cel-
lules. Au cours de l’infection, les cellules du système immunitaire
vont être activées et les DC vont présenter aux Lc T les Ag des agents
étrangers qui sont identiques à certains auto-Ag. Les Lc T activés
vont ensuite réagir contre les cellules exprimant ces Ag. Ainsi, le
sérum des patients infectés par le virus HTLV-1 responsable d’une
paraparésie spastique contient des taux élevés d’anticorps contre
un des Ag du virus HTLV-1, HTLV-1 tax. Ces anticorps reconnaissent
aussi un auto-Ag exprimé dans les neurones, la heterogenous nuclear
ribonuclear protein A-1 (protéine nucléaire hnRNP-A1). L’infection
virale a entraîné une présentation efficace des Ag viraux, ce qui a
déclenché une réponse immunitaire impliquant les Lc T et B abou-
tissant à la production d’autoanticorps qui vont être responsables
des lésions des neurones [9].
3.2.1.2. Adjuvants responsables d’inflammation. L’importance de la
présentation de l’Ag est illustrée par les MAI expérimentales. Dans
la plupart des modèles animaux utilisés, une protéine prove-
nant de l’organe contre lequel les chercheurs veulent déclencher
une MAI est injectée à l’animal, par exemple la myéline pour
induire une encéphalite auto-immune ou le cartilage pour induire
une polyarthrite. Néanmoins l’injection de la protéine seule ne
déclenche aucune réaction immunitaire. En revanche, l’injection
simultanée d’une protéine et d’un stimulant immunitaire entraî-
nant une inflammation (comme l’adjuvant complet de Freund
comprenant des huiles et du BCG, des lipopolysaccharides des bac-
téries de Gram négatif, des séquences CpG déméthylés des ADN
bactériens) induit chez certaines souches animales des MAI spé-
cifiques dirigées contre l’organe dont provient la protéine. Toutes
ces substances qui déclenchent une activation non spécifique de la
réponse immunitaire innée inflammatoire, sont appelés signaux
«danger ». Ces substances souvent d’origine infectieuse activent
les cellules de l’immunité innée (macrophages, polynucléaires, cel-
lules NK) en se liant à des récepteurs très bien conservés au cours
de l’évolution appelés toll-like receptor (TLR). L’inflammation non
spécifique entraîne l’attraction et l’activation de nombreuses cel-
lules de la réponse immunitaire innée mais aussi des cellules de
la réponse immunitaire adaptative qui soit expriment des TLR, soit
exprimentdes récepteurs auxchimiokines et cytokineslibérées. Les
DC jouent un rôle clé de lien entre la réponse innée et la réponse
adaptative. Les DC attirées sur le site inflammatoire captent les
Ag des protéines injectées, rec¸ oivent un signal d’activation et de
différenciation soit via leur TLR [10], soit via les cytokines inflam-
matoires et migrent au sein des ganglions au sein desquels elles
vont présenter efficacement des auto-Ag à des Lc T spécifiques.
3.2.1.3. Augmentation de l’activation et du nombre des cellules den-
dritiques. L’activation et l’augmentation du nombre de cellules
présentatrices d’Ag jouent probablement un rôle dans l’induction
des MAI. Cette hypothèse a été confirmée au cours du lupus éry-
thémateux disséminé [11]. Les DC peuvent dériver des monocytes
circulants qui se différencient soit en macrophages soit en DC
suivant le microenvironnement. En particulier, l’interféron (IFN)-
␣joue un rôle très important en induisant la différenciation des
monocytes en DC. Or le sérum de patients atteints de lupus contient
des taux élevés d’IFN-␣[12,13]. L’augmentation et l’activation du
nombre de DC est certainement un des facteurs importants dans
la physiopathogénie du lupus. Une des principales cellules produc-
trices d’IFN-␣est la cellule plasmacytoïde qui est un type particulier
de DC [14]. Du fait d’une susceptibilité génétique particulière,
les cellules plasmacytoïdes des patients lupiques produiraient des
quantités anormalement importantes d’IFN-␣après activation [15].
3.2.1.4. Modification de la balance cytokinique. Plusieurs cytokines
jouent un rôle clé dans l’activation de cellules immunitaires et une
augmentation de leur sécrétion pourrait être associées à certaines
MAI [16]. Ainsi, à titre d’exemple, les patients atteints de lupus
ont des taux élevés d’IL-10 sérique [17]. Cette hypersécrétion d’IL-
10 pourrait être d’origine environnementale [18] ; l’IL-10 stimule
les Lc B à produire des anticorps. Cette hyperstimulation chronique
est responsable de l’apparition d’autoanticorps.
3.2.2. Une dérégulation du système immunitaire peut entraîner
une maladie auto-immune
3.2.2.1. Rôle des lymphocytes T régulateurs (Treg). La découverte
des Lc Treg, Lc exprimant CD4, CD25, FoxP3 ayant des propriétés
immunosuppressives [19], a permis de mieux comprendre la phy-
siopathologie des MAI. Dans les modèles murins, la démonstration
de l’importance des Treg pour empêcher la survenue de MAI est
parfaitement claire [20]. Cependant chez l’homme, les publications
sur le rôle des Treg dans les MAI ont rapporté des nombreux résul-
tats discordants en partie expliqués par des problèmes techniques
de choix des anticorps et des tests fonctionnels. Il est probable que
les Treg jouent un rôle dans les MAI chez l’homme car la non-
fonctionnalité des Treg (par mutation de FoxP3) est responsable
de la survenue du syndrome IPEX (immune dysregulation, polyen-
docrinopathy, enteropathy et syndrome lié à l’X) qui associe une
sensibilité à des infections sévères et répétées, une polyendocri-
nopathie, une entéropathie et des lésions eczématiformes et des
affections auto-immunes (hypothyroïdie, anémie hémolytique et
thrombopénies) lors de la mutation de FoxP3, mais ce rôle est
probablement beaucoup moins important que dans les modèles
murins [21,22].
3.2.2.2. Anomalies du système du complément. Une anomalie au
niveau de la cascade des protéines du complément joue peut-être
un rôle dans la pathogénie de certaines MAI. Un des rôles majeurs
du complément est de favoriser l’épuration des corps apoptotiques
circulants provenant de la mort des cellules. Un dysfonctionne-
ment du complément entraîne la persistance de corps apoptotiques
en grandes quantités et ces corps apoptotiques sont des auto-Ag
potentiels qui peuvent ensuite être présentés aux Lc T et déclencher
des MAI. Ainsi des déficits en fractions C1, C2 et C4 sont fréquem-
ment retrouvés chez les patients atteints de lupus systémique [23].
4. Conclusion
De nombreux travaux utilisant des modèles murins et chez
l’homme ont permis de mieux appréhender la physiopathologie
des MAI. Les MAI surviennent lorsque les auto-Ag, du fait d’une
infection virale, d’un contexte inflammatoire, d’une anomalie des
systèmes de régulation, sont présentés par des cellules présenta-
trices d’Ag qui vont ainsi pouvoir stimuler les Lc autoréactifs. Même
si la connaissance actuelle des mécanismes physiopathologiques
divers impliquant des facteurs génétiques et des facteurs environ-
nementaux reste encore imparfaite, elle a permis le développement
de nouvelles voies thérapeutiques.

B. Bonnotte / La Revue de médecine interne 31S (2010) S292–S295 S295
Conflit d’intérêt
Pas de conflit d’intérêt.
Références
[1] Austrup F, Vestweber D, Borges E, et al. P- and E-selectin mediate recruit-
ment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature
1997;385:81–3.
[2] von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the
same coin. N Engl J Med 2000;343:1020–34.
[3] Klein J, Sato A. The HLA system. Second of two parts. N Engl J Med
2000;343:782–6.
[4] Henderson RD, Bain CJ, Pender MP. The occurrence of autoimmune diseases
in patients with multiple sclerosis and their families. J Clin Neurosci
2000;7:434–7.
[5] Ginn LR, Lin JP, Plotz PH, et al. Familial autoimmunity in pedigrees of idiopa-
thic inflammatory myopathy patients suggests common genetic risk factors for
many autoimmune diseases. Arthritis Rheum 1998;41:400–5.
[6] Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med
2008;358:929–39.
[7] Sospedra M, Ferrer-Francesch X, Dominguez O, Juan M, Foz-Sala M, Pujol-
Borrell R. Transcription of a broad range of self-antigens in human thymus
suggests a role for central mechanisms in tolerance toward peripheral antigens.
J Immunol 1998;161:5918–29.
[8] Ohashi PS, Oehen S, Buerki K, et al. Ablation of “tolerance” and induction of dia-
betes by virus infection in viral antigen transgenic mice. Cell 1991;65:305–17.
[9] Levin MC, Lee SM, Kalume F, et al. Autoimmunity due to molecular mimicry as
a cause of neurological disease. Nat Med 2002;8:509–13.
[10] Hurst J, von Landenberg P. Toll-like receptors and autoimmunity. Autoimmun
Rev 2008;7:204–8.
[11] Crispin JC, Alcocer-Varela J. The role myeloid dendritic cells play in the
pathogenesis of systemic lupus erythematosus. Autoimmun Rev 2007;6:
450–6.
[12] Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signa-
tures in systemic lupus erythematosus blood. J Exp Med 2003;197:711–
23.
[13] Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendri-
tic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science
2001;294:1540–3.
[14] Lehmann P, Homey B. Clinic and pathophysiology of photosensitivity in lupus
erythematosus. Autoimmun Rev 2009;8:456–61.
[15] Jin O, Kavikondala S, Mok MY, et al. Abnormalities in circulating plasmacytoid
dendritic cells in patients with systemic lupus erythematosus. Arthritis Res
Ther 2010;12:R137.
[16] Mok MY, Wu HJ, Lo Y, Lau CS. The relation of interleukin17 (IL-17) and IL-
23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus.
J Rheumatol 2010;37:2046–52.
[17] Llorente L, Richaud-Patin Y, Couderc J, et al. Dysregulation of interleukin-
10 production in relatives of patients with systemic lupus erythematosus.
Arthritis Rheum 1997;40:1429–35.
[18] Grondal G, Kristjansdottir H, Gunnlaugsdottir B, et al. Increased number of
interleukin-10-producing cells in systemic lupus erythematosus patients and
their first-degree relatives and spouses in Icelandic multicase families. Arthritis
Rheum 1999;42:1649–54.
[19] Taguchi O, Nishizuka Y. Self tolerance and localized autoimmunity. Mouse
models of autoimmune disease that suggest tissue-specific suppressor T cells
are involved in self tolerance. J Exp Med 1987;165:146–56.
[20] Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat
Rev Immunol 2002;2:389–400.
[21] Kukreja A, Cost G, Marker J, et al. Multiple immuno-regulatory defects in type-
1 diabetes. J Clin Invest 2002;109:131–40.
[22] Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Iso-
lation and functional characterization of regulatory CD25 bright CD4+ T cells
from the target organ of patients with rheumatoid arthritis. Eur J Immunol
2003;33:215–23.
[23] Hartmann D, Fremeaux-Bacchi V, Weiss L, et al. Combined heterozygous defi-
ciency of the classical complement pathway proteins C2 and C4. J Clin Immunol
1997;17:176–84.
1
/
4
100%