Fascicule de TP

Université Claude Bernard – Lyon I
Travaux pratiques de thermodynamique
Module thermodynamique II
L3 – parcours physique
2013-2014

• T.P. n°1 : Changement d’état d’un corps pur
• T.P. n°2: Changement d’état d’un système binaire
• T.P. n°3: Moteur de Stirling
• T.P. n°4: Chaleurs spécifiques des gaz parfaits et Détente de
Joule-Thomson


T.P. n°1
Changements d’état d’un corps pur

TP n°1 : Changements d’état d’un corps pur
TP 1 – page 2
T.P. n°1
Changement d’état d’un corps pur
I. Objectifs de la manipulation
On se propose ici d’étudier le changement d’état d’un corps pur. Deux types de transitions de
phase seront étudiés dans ce TP :
L’équilibre liquide-gaz
L’équilibre liquide-solide.
Le passage de l’état liquide à l’état gazeux va être effectué par compression ou détente
isotherme d’un fluide (SF6). On va pouvoir déterminer expérimentalement les isothermes
d’Andrews. Le premier objectif de ce TP sera donc de réaliser la compression et la
liquéfaction d’un gaz au voisinage du point critique et d’exploiter aussi complètement que
possible les renseignements donnés par la connaissance des paramètres d’état en tout point de
l’espace P, V, T.
L’étude de l’équilibre liquide-solide sera réalisée par le chauffage d’un corps pur (salol ou
étain). En mesurant l’évolution au cours du temps de la température de ce corps, il est
possible d’obtenir des informations sur la fusion et de la solidification de ce corps. La notion
de surfusion sera également abordée.
II. Rappels sur la description thermodynamique d’un corps pur
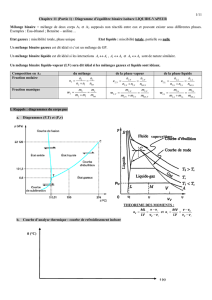
1) Isothermes d’un fluide pur - diagramme de CLAPEYRON
Un corps pur est décrit par des variables mécaniques
(pression P, volume V) et thermique (Température T). Ces
paramètres sont liés par l’équation d’état :
(,,) 0FPVT
(1)
Selon les conditions expérimentales, apparaissent l’une ou
l’autre des 3 phases : gaz, liquide, solide, ou bien un
mélange de ces phases.
Restreignons-nous au cas où le fluide est gazeux, ou
liquide, ou un mélange de ces 2 phases. La représentation
la plus couramment adoptée pour le décrire est le
diagramme de CLAPEYRON, qui est une coupe à T
constant, de la surface d’équation (1).
Deux types d’isothermes apparaissent :
Pour des températures supérieures à une
température Tc dite “ Température critique ”, le
fluide est gazeux. Toute réduction de son volume
s’accompagne d’une augmentation de pression. La
loi de compression est approximativement la loi de
MARIOTTE. Pour une mole : PV RT (2)
Où R est la constante des gaz parfaits.
Pour des températures inférieures à la température
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
1
/
54
100%