Cerezyme® : Instructions d`utilisation – reconstitution, dilution et

Cerezyme® (imiglucérase) est indiqué pour le traitement de substitution enzymatique de longue durée des patients ayant reçu un diagnostic confirmé de la maladie de Gaucher non neuronopathique (type 1) ou
neuronopathique chronique (type 3), qui présentent des symptômes non neurologiques cliniquement significatifs de la maladie.
Les symptômes non neurologiques de la maladie de Gaucher peuvent comprendre une ou plusieurs des affections suivantes :
- anémie après exclusion d’autres causes, par exemple un déficit en fer
- thrombocytopénie
- maladie osseuse après exclusion d’autres causes, telles qu’une carence en vitamine D
- hépatomégalie ou splénomégalie
Les informations suivantes sont destinées exclusivement aux médecins ou à d’autres professionnels de la santé : Instructions d’utilisation – reconstitution, dilution et administration
Respectez les règles d’axénie (pas
de germes)
Préparation
1. Les flacons doivent être conservés au réfrigérateur à une
température comprise entre 2 °C et 8 °C.
2. Préparez le matériel :
• Le nombre de flacons de Cerezyme requis est
déterminé d’après le poids du patient. Chaque flacon
contient 400 unités d’imiglucérase. Les flacons
être retirés du réfrigérateur environ 30 minutes avant
la préparation afin d’être amenés à température
ambiante. Vérifiez la date de péremption imprimée sur
le fond de l’emballage du flacon (n’utilisez pas
Cerezyme® après la date d’expiration).
• Eau stérile pour préparations injectables pour
reconstituer Cerezyme®
• Solution de chlorure de sodium à 0,9 % pour
administration intraveineuse, 2 x 100 ml
• Solution de chlorure de sodium à 0,9 % pour
administration intraveineuse, 2 x 50 ml pour rincer la
tubulure de perfusion avant et après la perfusion
• Chlorhexidine à 0,5 % en solution alcoolique à 70 %
(solution antiseptique)
• Nombre correct de seringues de 10 ml et 50 ml, selon
la dose de Cerezyme
• 3 aiguilles hypodermiques stériles (1,1 × 40 mm) ;
1 aiguille à ailettes
• Filtre en ligne de 0,2 µm à faible fixation protéinique
• Plateau pour aiguilles hypodermiques, ruban
Micropore, Mediswabs (tampons imbibés d’alcool),
conteneur à aiguilles, nettoyant pour les mains
Reconstitution
3. Retirez la capsule amovible du flacon de Cerezyme.
4. Désinfectez le bouchon en caoutchouc du flacon de
Cerezyme à la chlorhexidine et laissez sécher à l’air libre
5. Ouvrez l’eau pour préparations injectables
6. Aspirez le nombre requis de ml d’eau stérile pour
préparations injectables dans la seringue : 10,2 ml pour
des flacons de 400 unités.
7. Injectez l’eau pour préparations injectables lentement
contre la paroi en verre de chaque flacon.
8. Remuez le ou les flacons soigneusement pour mélanger
la solution (évitez de secouer vigoureusement la solution
reconstituée afin d’éviter la formation de mousse).
9. De petites bulles d’air pourraient apparaître après le
mélange.
10. Laissez reposer la solution pendant quelques minutes
pour faire disparaître les éventuelles bulles d’air et
garantir que la poudre soit correctement reconstituée
(vérifiez l’absence de particules étrangères ou d’une
coloration anormale de la solution).
Dilution
11. Désinfectez le capuchon/l’ouverture de 1 ou 2 poches de
solution de chlorure de sodium à 0,9 % pour
administration intraveineuse avec de la chlorhexidine et
laissez sécher à l’air libre.
12. Calculez la quantité de solution reconstituée de
Cerezyme® qui est présente dans les flacons et prélevez
la même quantité dans la poche contenant la solution de
chlorure de sodium. Il y aura alors suffisamment
d’espace pour ajouter la solution reconstituée de
Cerezyme.
Par exemple, si la quantité prescrite est de 3 flacons de
Cerezyme® de 400 unités chacun, retirez alors 30 ml
(= 3 x 10 ml) de solution de chlorure de sodium de la
poche de solution de chlorure de sodium. Ne retirez
jamais plus de la moitié du contenu de la poche de
solution de chlorure de sodium afin d’être sûr que la
solution diluée se compose pour moitié au moins de
chlorure de sodium.
13. À l’aide d’une ou de plusieurs seringues de 50 ml,
prélevez 10 ml (flacon de 400 unités) dans les flacons.
Au moment du prélèvement de ces quantités, le produit
ne doit pas contenir de mousse.
Injectez ensuite lentement le volume total de solution de
Cerezyme® dans la poche de solution de chlorure de
sodium à 0,9 %.
14. Mélangez cette solution de Cerezyme avec précaution.
15. Pendant l’administration, la solution reconstituée doit
être filtrée par un filtre en ligne de 0,2 µm à faible pouvoir
de fixation protéique.
Administration
16. La dose et la vitesse de perfusion de Cerezyme® seront
fixées par le médecin traitant.
17. Cerezyme® doit être administré par perfusion
intraveineuse.
18. La solution doit être administrée dans les trois heures de
la reconstitution.
19. Avant la perfusion, remplissez le système de perfusion
avec la solution de Cerezyme® et retirez toutes les bulles
d’air présentes.
20. Pour s’assurer que la dose de traitement totale est
administrée, après la fin de la perfusion, rincez la
tubulure avec une poche de chlorure de sodium à 0,9 %
pour administration intraveineuse de 50 ml sans
augmenter la vitesse de perfusion.
21. Du point de vue de la sécurité microbiologique, la
préparation doit être utilisée immédiatement. Si la
préparation ne peut être utilisée immédiatement, elle
peut être conservée pendant un maximum de 24 heures
au réfrigérateur, à une température comprise entre 2 °C
et 8 °C et à l’abri de la lumière.
Détails en cas d’urgence (à compléter par le médecin traitant)
Actions en cas de réaction grave liée à la perfusion
1. ARRÊTEZ immédiatement la perfusion
2. Appelez le numéro des urgences (100 ou 112)
Appelez le médecin au .........
Pour tous les effets indésirables, consultez la rubrique 4.8 du RCP au verso de
cette affiche.
Si vous présentez un effet indésirable, avertissez IMMÉDIATEMENT le
médecin traitant. Le patient devra peut-être recevoir d’autres médicaments pour
éviter une réaction allergique (p. ex. des antihistaminiques et/ou des
corticostéroïdes).
Traitement à domicile
• Le domicile doit être adapté à un traitement par perfusion et doit notamment : être propre, disposer
de l’électricité et de l’eau courante, d’une ligne téléphonique, d’un réfrigérateur et d’un espace
physique pour conserver Cerezyme® et d’autres fournitures nécessaires à la perfusion.
• Il est préférable qu’une personne soignante/un tiers soit présent(e) auprès du patient.
• Le patient et/ou la personne soignante ont suivi la formation appropriée sur les procédures de
reconstitution, de dilution et de perfusion de Cerezyme.
• Un système de perfusion portable, tel qu’un diffuseur portable, peut être utilisé (système de
perfusion à pression positive).
• Un kit permettant de traiter un éventuel choc anaphylactique doit toujours être disponible.
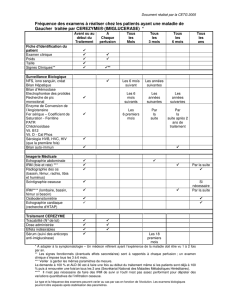
Étape 13 : lorsque le produit est
prélevé, il ne peut pas contenir de
mousse
Étape 12 : prélevez 10 ml
(flacon de 400 U) dans la poche
pour chaque flacon utilisé et
jetez-la
Étape 8 : tournez prudemment le
flacon en un mouvement circulaire
avec les mains
Étape 7 :
injectez l’eau pour préparations
injectables
Étape 6 :
prélevez l’eau stérile
Étape 2 :
préparez le matériel
Les autorités sanitaires européennes ont fixé des conditions à la mise sur le marché du médicament CEREZYME®. Le
plan obligatoire de réduction des risques en Belgique, dont ces informations font partie, est une mesure prise pour
garantir un usage sûr et efficace de CEREZYME® (version RMA 07/2014).
Cerezyme® : Instructions d’utilisation – reconstitution, dilution et administration
– Affiche destinée au médecin et à l’équipe soignante du patient –
Lisez attentivement le RCP (Résumé des caractéristiques du produit) au verso avant de prescrire Cerezyme® . Les
professionnels de la santé sont priés de signaler les effets indésirables de Cerezyme® au Centre Belge de
Pharmacovigilance pour les médicaments à usage Humain (CBPH). Consultez le Guide destiné aux professionnels de la
santé.

1.
DÉNOMINATION DU MÉDICAMENT
Cerezyme 400 U poudre pour solution à diluer pour perfusion
2.
COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 400 unités* d’imiglucérase**.
Après reconstitution, la solution contient 40 unités (environ 1,0 mg) d’imiglucérase par ml
(400 U/10 ml).
* Une unité enzymatique (U) est définie comme la quantité d’enzyme qui catalyse l’hydrolyse
d’une micromole du substrat synthétique para-nitrophényl--D-glucopyranoside (pNP-Glc) en une
minute à 37 °C.
** L’imiglucérase est une forme modifiée de la -glucosidase acide humaine ; c’est une protéine
obtenue par la technologie de l’ADN recombinant à partir d’une culture de cellules ovariennes de
hamsters chinois, avec modification du mannose afin de cibler les macrophages.
Excipients :
Pour la liste complète des excipients, voir rubrique 6.1.
Ce médicament contient du sodium administré sous forme de solution intraveineuse de chlorure
de sodium à 0,9 % (voir rubrique 6.6). Après reconstitution, la solution contient 1,24 mmol de
sodium (400 U/10 ml). Les patients suivant un régime contrôlé en sodium doivent en tenir compte.
3.
FORME PHARMACEUTIQUE
Poudre pour solution à diluer pour perfusion
Cerezyme est une poudre blanche à blanchâtre.
4.
DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Cerezyme (imiglucérase) est indiqué pour le traitement de substitution enzymatique de longue
durée des patients ayant reçu un diagnostic confirmé de la maladie de Gaucher non
neuronopathique (type 1) ou neuronopathique chronique (type 3), qui présentent des symptômes
non neurologiques cliniquement significatifs de la maladie.
Les symptômes non neurologiques de la maladie de Gaucher peuvent comprendre une ou
plusieurs des affections suivantes :
• anémie après exclusion d’autres causes, par exemple un déficit en fer
• thrombocytopéniemaladie osseuse après exclusion d’autres causes, telles qu’une carence
en vitamine D
• hépatomégalie ou splénomégalie
4.2 Posologie et mode d’administration
La prise en charge de la maladie doit se faire sous la supervision de médecins qui connaissent le
traitement de la maladie de Gaucher.
Posologie
Vu la nature hétérogène et multisystémique de la maladie de Gaucher, la posologie doit être
adaptée à chaque patient, après une évaluation approfondie de tous les symptômes cliniques de
la maladie. Dès que la réponse individuelle du patient concernant tous les symptômes cliniques
pertinents a été clairement établie, les doses et fréquences auxquelles le produit est administré
peuvent être adaptées afin de maintenir les paramètres optimaux déjà obtenus pour tous les
symptômes cliniques ou d’améliorer les paramètres cliniques qui ne sont pas encore normalisés.
Différents schémas posologiques se sont avérés efficaces pour certains ou tous les symptômes
non neurologiques. Une dose initiale de 60 U/kg de poids corporel une fois toutes les deux
semaines a permis d’observer, en six mois de traitement, une amélioration des paramètres
hématologiques et viscéraux ; la poursuite du traitement a permis d’enrayer la progression de la
maladie osseuse ou d’améliorer la maladie osseuse. L’administration de faibles doses de
seulement 15 U/kg de poids corporel une fois toutes les deux semaines a mis en évidence une
amélioration des paramètres hématologiques et de l’organomégalie, mais pas des paramètres
osseux. La fréquence de perfusion habituelle est d’une fois toutes les deux semaines ; la plupart
des données disponibles porte sur cette fréquence de perfusion.
Patients pédiatriques
La posologie ne doit pas être adaptée pour les patients pédiatriques.
L’efficacité de Cerezyme sur les symptômes neurologiques des patients atteints de la forme
neuronopathique chronique de la maladie de Gaucher n’est pas établie et aucun schéma
posologique spécial ne peut être recommandé pour ces symptômes (voir rubrique 5.1).
Mode d’administration
Après reconstitution et dilution, la préparation est administrée par perfusion intraveineuse. Au
départ, la vitesse à laquelle Cerezyme est administré ne peut excéder 0,5 unité par kg de poids
corporel par minute. Lors des perfusions suivantes, la vitesse de perfusion peut être augmentée,
mais jamais au-delà de 1 unité par kg de poids corporel par minute. L’augmentation de la vitesse
de perfusion doit se faire sous la supervision d’un professionnel de la santé au centre de soins de
santé.
La perfusion à domicile de Cerezyme peut être envisagée pour les patients ayant bien toléré leurs
perfusions pendant plusieurs mois. La décision de faire passer le patient à la perfusion à domicile
doit être prise après évaluation et recommandation par le médecin traitant. Avant de pouvoir
commencer les perfusions de Cerezyme à domicile par le patient ou la personne soignante, un
médecin ou un(e) infirmier(ère) doit former le patient ou la personne soignante en milieu clinique.
Le patient ou la personne soignante reçoit des instructions relatives à la technique de perfusion et
à la tenue du journal du traitement. Les patients qui développent des effets indésirables pendant
la perfusion doivent immédiatement arrêter la perfusion et prendre contact avec un médecin ou
un(e) infirmier(ère) et les éventuelles perfusions suivantes doivent avoir lieu en milieu clinique. La
dose et la vitesse de perfusion doivent rester constantes lors de l’administration à domicile et ne
peuvent pas être modifiées sans l’accord d’un médecin ou d’un(e) infirmier(ère).
Pour les instructions concernant la reconstitution et la dilution du médicament avant
l’administration, consultez la rubrique 6.6.
Les médecins et autres professionnels de la santé sont encouragés à inscrire les patients atteints
de la maladie de Gaucher, ainsi que les patients présentant des symptômes neuronopathiques
chroniques de la maladie, dans le « Registre ICGG de la maladie de Gaucher » (voir
rubrique 5.1).
4.3 Contre-indications
Hypersensibilité au principe actif ou à l’un des excipients (voir rubrique 4.4).
4.8 Effets indésirables
Dans le tableau ci-dessous, les effets indésirables sont présentés par classes de systèmes
d’organes et par fréquence (fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100) et rare
(≥ 1/10 000 à < 1/1 000)). Dans chaque groupe de fréquence, les effets indésirables sont classés
par ordre décroissant de gravité.
Affections du système nerveux
Peu fréquent :
Vertiges, maux de têtes, paresthésies*
Affections cardiaques
Peu fréquent :
Tachycardie*, cyanose*
Affections vasculaires
Peu fréquent :
Rougeur du visage*, hypotension*
Affections respiratoires, thoraciques et médiastinales
Fréquent :
Dyspnée*, toux*
Affections gastro-intestinales
Peu fréquent :
Vomissements, nausées, crampes abdominales, diarrhée
Affections du système immunitaire
Fréquent :
Rare :
Réactions d’hypersensibilité
Réactions anaphylactoïdes
Affections de la peau et du tissu sous-cutané
Fréquent :
Urticaire/angio-œdème*, prurit*, éruption*
Affections musculo-squelettiques et systémiques
Peu fréquent :
Arthralgie, mal de dos*
Troubles généraux et anomalies au site d’administration
Peu fréquent :
Irritation, sensation de brûlure, œdème, abcès aseptique sur le
site de perfusion, sensation d’oppression thoracique*, fièvre,
frissons, fatigue
Un total d’environ 3 % des patients présentent des symptômes indiquant une hypersensibilité
(signalés par un * dans le tableau ci-dessus). Ces symptômes sont apparus pour la première fois
pendant les perfusions ou peu après. Ces symptômes réagissent généralement bien à un
traitement par antihistaminiques et/ou corticostéroïdes. Il faut avertir les patients qu’ils doivent
arrêter la perfusion du produit et prendre contact avec leur médecin si ces symptômes
apparaissent.
7.
TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Genzyme Europe B.V., Gooimeer 10, 1411 DD, Naarden, Pays-Bas.
8.
NUMÉROS DE L’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/97/053/003
EU/1/97/053/004
EU/1/97/053/005
9.
DATE DE PREMIÈRE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
Date de première autorisation : 17 novembre 1997
Date du dernier renouvellement : 17 septembre 2007
10.
DATE DE MISE À JOUR DU TEXTE
10/2010
Des informations détaillées sur ce produit sont disponibles sur le site internet de l’Agence
européenne des médicaments http://www.ema.europa.eu/.
1
/
2
100%