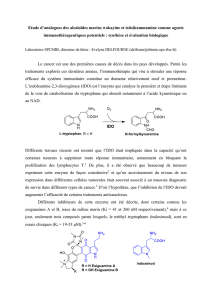
Synthèse de molécules d`intérêt biologique: Préparation des dérivés

R
ÉPUBLIQUE
A
LGÉRIENNE
D
ÉMOCRATIQUE ET
P
OPULAIRE
M
INISTÈRE
DE
L’
ENSEIGNEMENT
SUPÉRIEUR ET DE LA
R
ECHERCHE
S
CIENTIFIQUE
Université Mentouri-Constantine
Faculté des Sciences Exactes
Département de Chimie
N° d’ordre………
Série…………….
THÈSE
PRÉSENTÉE AU DÉPARTEMENT DE CHIMIE EN VUE
DE L’OBTENTION DU DIPLÔME DE DOCTORAT EN SCIENCES
OPTION
SYNTHÈSE ORGANIQUE
THÈME
Synthèse de molécules d’intérêt biologique:
Préparation des dérivés de la 3,4-dihydropyrimidinone
et de la 1,4-dihydropyridine par des réactions à
composants multiples
PAR
Wassima GHALEM
Devant le Jury:
P
résident S. RHOUATI Prof Univ. Mentouri Constantine
R
apporteur A. DEBACHE Prof Univ. Mentouri Constantine
E
xaminateurs N. AOUF Prof Univ.Annaba Annaba
A. GOUASMIA Prof Univ. Tebessa Tebessa
L. BOUDIBA
B. BOUMOUD
M.C
M.C
Univ. Tebessa
Univ. Mentouri
Tebessa
Constantine


Avant-propos
Les travaux décrits dans ce manuscrit ont été effectués au sein du Laboratoire des Produits
Naturels d’Origine Végétale et de Synthèse Organique du département de chimie de la faculté des
sciences Exactes de l’université Mentouri-Constantine, dirigé par le proffeseur A. Debache.
Je tiens tout particulièrement à lui exprimer ma profonde reconnaissance pour m’avoir
accueillie au sein de son équipe et pour toute la confiance qu’il m’a accordée tout au long de ce
travail. J’aimerais aussi le remercier pour l’intérêt qu’il a porté à mes travaux et pour sa passion
pour la Synthèse Organique qu’il m’a fait partager. Je tiens à lui témoigner toute ma gratitude
pour tous les conseils, orientations et encouragements qu’il n’a cessés de me prodiguer tout au
long de la réalisation de ce travail.
J’adresse mes mes remerciements à monsieur S. Rhouati, professeur à l’université Mentouri-
Constantine, pour avoir accepté de présider le jury de soutenance.
Je tiens aussi à remercier, monsieur N. Aouf, professeur à l’université d’Annaba, monsieur A.
Gouasmia, professeur à l’université de Tebessa, madame L. Boudiba maître de conférences à
l’université de Tebessa, monsieur B. Boumoud maître de conférences à l’université Mentouri-
Constantine, pour avoir accepté de juger ce travail.
Je remercie chaleureusement tout les membres de ma promotion: Belbache, Belloum,
Bensuisi, Boumaaraf, Chebbah, Tiba, Charwana, Saouda, Boutiti, kitouni, qui ont rendu très
agréables ces quelques années passées en leur compagnie.
Mes remerciements s’adressent également à Messieurs A. Haichour responsable de la
spectrométrie de résonance magnétique nucléaire et A. Benhamoud chargé de la spectroscopie
infra-rouge pour leur disponibilité.
Un grand merci à tous mes collègues de laboratoire PHYSYNOR étudiants et enseignants
particulièrement à Amimour, Tafer, Benzarka, Choughiat, Boulcina et Bouraiou. Merci aussi à
Sara, Imen, Amel ahmed. Mes remerciements s’adressent également à Zahra pour son amitié et sa
présence.

Sommaire
INTRODUCTION GENERALE ...………………………………………………………
Bibliographie………………………………………………………………………………
01
04
Chapitre I : NOUVELLE VOIE D’ACCES AUX PRODUIT DE BIGINELLI
I- La réaction de Biginelli……………………………………………………………………..
I-1-Réactivités des dihydropyrimidinones……………………………………………..
I-2- Intérêts biologiques………………………………………………………………..
II- Étude et analyse de la réaction de Biginelli……………………………………………….
II-1- Les différents composants de la réaction de Biginelli…………………………….
II-2- La catalyse………………………………………………………………………..
II-3-Le solvant…………………………………………………………………………..
II-4-Le mécanisme………………………………………………………………………
II-5- Autres méthodes…………………………………………………………………..
III- Résultats et discussions………………………………………………………………….
III-1- optimisation des conditions………………………………………………………
III-2-Généralisation de la réaction de Biginelli catalysée par Et3N……………………
III-3- Étude du mécanisme…………………………………………………………….
III-4- Étude Spéctrale………………………………………………………………….
IV- Conclusion……………………………………………………………………………….
VI- Partie expérimentale………………………………………………………………………
Bibliographie………………………………………………………………………………….
05
06
08
12
12
15
27
27
30
32
33
34
35
36
43
44
51

Chapitre II : DÉVELOPPEMENT DE NOUVEAUX CATALYSEURS DE LA
RÉACTION DE HANTZSCH
I- La réaction de Hantzsch……………………………………………………………..
I-1-Laréactivité des 1,4-DHPs……………………………………………………..
I-2- Intérêts biologiques…………………………………………………………..
II-Etude de la réaction de Hantzsch ……………………………………………………
II-1-Les acides de Lewis…………………………………………………………..
II-2- Les supports solides…………………………………………………………..
II-3- Les hétéropolyacides………………………………………………………….
II-4- Les liquides ioniques………………………………………………………….
II-5- Le micro-onde………………………………………………………………..
II-6- Autres catalyseurs…………………………………………………………….
II-7-Autre méthodes………………………………………………………………..
III-Résultats et discussion……………………………………………………………….
III-1- Optimisation des conditions………………………………………………….
III-2- généralisation de la réaction………………………………………………….
III-3- Étude du mécanisme…………………………………………………………
III-4- Étude spéctrale……………………………………………………………….
IV-Conclusion…………………………………………………………………………..
V-Partie expérimentale…………………………………………………………………
Bibliographie……………………………………………………………………………
CONCLUSION GENERALE……………………………………………………………
ANNEXE 1
ANNEXE 2
59
59
62
67
71
71
72
73
74
75
76
78
78
80
83
84
91
92
103
108
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
1
/
135
100%