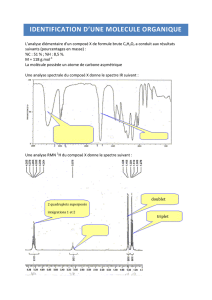
Table des matières Chapitre I Photométrie de flamme

/méthodes spectroscopiques d’analyse/
Table des matières
Chapitre I
Photométrie de flamme…………………………………….. page 1
Chapitre II
Absorption atomique ………………………………………. page 5
Chapitre III Spectrophotométrie UV-Visible ……………………………page11
Chapitre IV
Spectrophotométrie d’absorption dans l’infrarouge……...page 23
Chapitre V Spectroscopie de résonance magnétique nucléaire du proton (RMN1H)
.…………………………………………………………………page 38
Chapitre VI
Spectrométrie de masse ………………………………………page 49

/méthodes spectroscopiques d’analyse/
Photométrie de flamme
I) Introduction
Le phénomène d’émission de radiations par des substances excitées sont extrêmement variés
à la fin par la nature des excitations et du domaine de longueur d’onde considérée.
La photométrie de flamme est une technique d’analyse qui se base sur l’émission.
II) Principe
Dans la photométrie de flamme, l’excitation est faite à l’aide d’une flamme ou avec
l’élémentà doser. Après avoir été sélectionnées, les radiations sont évaluées en fonction de
leurs intensités lesquelles sont proportionnelles aux nombres d’atomes excités donc à la
concentration totale de l’élément présent dans la flamme.
La flamme est connue comme étant un moyen d’excitation peu puissant. Ceci entrainera
deux conséquences importantes :
-le spectre obtenu sera peu fourni en raies
-les éléments à bas potentiel d’excitation seront les seules à être excités : Alcalins, Alcalino-
terreux et parfois l’aluminium. Les plus importants sont les alcalins.
III) Phénomènes produits dans la flamme
Lorsque la solution arrive dans la flamme il se produit :
a) Une évaporation du solvant
b) Une fusion et une vaporisation des sels dissous
c) Une dissociation des molécules
d) Excitation des atomes
Il importe de prendre en considération le point de fusion du sel considéré ainsi que son
point d’ébullition.
Exemple : NaCl son point de fusion est 830°C, son point d’ébullition est 1480°C, il sera
vaporisé dans une flamme ordinaire
Pour le CaO son point de fusion est 2580°C donc il exigera une flamme de 3000°C
environ. Donc une flamme du type Oxy-acetylénique ( 3100°C).

/méthodes spectroscopiques d’analyse/
IV)Appareillage
Schéma :
7 4 5 6
flamme
3 filtre détecteur
alimentation enregistreur
en carburant
2
Alimentation capillaire
en comburant
1
Schéma 1 : Spectromètre de flamme
1)-solution à analyser par un capillaire
2)-pulvérisateur
3)-bruleur
4)-sélectionneur de longueur d’onde
5)-détecteur
6)-enregistreur
7)-flamme
V) Application analytique
Par la photométrie de flamme on dose surtout les alcalins( Na, K, Li).
1) Dosage directe : on rappelle que pour une température de flamme donnée l’intensité de
la radiation émise est proportionnelle à la concentration. Les principales étapes sont :
*on prépare une gamme de solutions obtenues par dilution d’une solution mère
*régler le zéro électrique

/méthodes spectroscopiques d’analyse/
*placer le filtre pour sélectionnée la longueur d’onde :pour Na on emploiera un filtre à
589nm et pour le K un filtre à 767nm
* régler la sensibilité en avancent l’aiguille à la valeur maximum et ce en pulvérisant la
solution la plus concentrée.
*faire passer les solutions de la gamme et tracer la courbe d’étalonnage.
2) méthode de l’étalon interne
Ajouté à chaque échantillon une quantité connue et identique d’un élément autre que celui à doser(
Li dans le dosage du K et Na). On mesure le rapport des intensités de la raie analysé et de la raie
étalon, se reporter ensuite à la courbe d’étalonnage pour la détermination de la concentration de la
solution à analyser.
Les étalons doivent être purs.
3)photométrie de flamme comparée aux méthodes chimiques
-Avantages : Réalisation de dosage en série, dosage de plusieurs métaux dans un mélange , grande
sensibilité de la méthode qui permet de travailler aux grandes dilutions.
-Inconvénients : limitation à certains cations, appareillage relativement couteux et problèmes
d’interférences qu’il n’est pas possible d’éliminer.
VI) Comparaison entre photométrie de flamme et absorption atomique
-en photométrie par émission ce sont les atomes excités qui émettent les radiations en revenant à
l’état fondamental et c’est ensuite l’intensité des radiations qui est mesurée.
-en absorption atomique ce sont les atomes à l’état fondamental qui absorbent la radiation de la
résonnance et c’est ensuite le pourcentage d’absorption qui sera mesuré
Le principal avantage de l’absorption atomique sur la photométrie par émission découle de
ce que l’absorption est fonction du nombre d’atomes (flamme large) à l’état fondamental ,
alors en émission il est fonction du nombre d’atomes excités ( flamme étroite).Ceci a pour
résultat l’application de la méthode par absorption atomique a un bien plus grand nombre
d’élément que dans le cas de l’émission.
VI) Interférence en photométrie de flamme
Leurs caractéristique est de gêner le dosage par divers interactions

/méthodes spectroscopiques d’analyse/
1) Chimiques : ce sont les effets produits par l’ensemble des substances dissoutes
Dans le cas des anions, les interactions sont négligeables en milieu dilué. Mais aux
concentrations supérieures à 0.1M, les anions Po4-3 , NO 3-et SO4-2 diminuent
l’intensité d’émission. Tel est l’effet des phosphates sur l’émission du calcium . Le
phénomène parait lié à la formation d’un composé stable
Ca3 (Po4)2 (dans ce cas) à la température de flamme considérée. Ainsi on observe une
réduction du nombre d’atomes de calcium susceptible d’être excité.
La présence de certains cations peut provoquer une absorption relativement forte du
rayonnement ce qui va diminuer considérablement la sensibilité de l’élément à doser.
2) interaction d’ionisation : l’ionisation est un phénomène parasite, elle augmente avec la
température. Le but recherché est l’excitation seulement en analyse par émission. Pour palier à cet
inconvénient on a donc avantage à utiliser des flammes relativement peu chaudes et des solutions
dilué
3) Facteurs physiques : l’augmentation de la viscosité de la solution se traduit par une diminution
de l’émission due à une mauvaise vaporisation. Donc l’influence du solvant est importante en
analyse par émission.
4) Concentration : en analyse par émission l’intensité I varie linéairement avec la concentration C .
En milieu plus concentré la linéarité n’est plus respectée.
Absorption atomique
I) Introduction
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
1
/
65
100%