Godin-Ethier_Jessica_2010_these

Université de Montréal
Contrôle de la réponse immunitaire par
l’indoleamine 2,3-dioxygénase: étude de la régulation
d’une molécule immunosuppressive dans les cellules
cancéreuses et les lymphocytes B chez l’humain
par
Jessica Godin-Ethier
Programme de Sciences Biomédicales
Faculté de Médecine
Thèse présentée à la Faculté de Médecine
en vue de l’obtention du grade de Doctorat
en Sciences Biomédicales
Août 2010
© Jessica Godin-Ethier, 2010

Université de Montréal
Faculté des études supérieures et postdoctorales
Cette thèse intitulée :
Contrôle de la réponse immunitaire par l’indoleamine 2,3-dioxygénase:
étude de la régulation d’une molécule immuno-suppressive dans les cellules cancéreuses et
les lymphocytes B chez l’humain
Présentée par :
Jessica Godin-Ethier
a été évaluée par un jury composé des personnes suivantes :
Dr Jean-François Gauchat, président-rapporteur
Dr Réjean Lapointe, directeur de recherche
Dr Hugo Soudeyns, membre du jury
Dr Frank Housseau, examinateur externe
Dr Gerardo Ferbeyre, représentant du doyen de la Faculté de Médecine

i
Résumé
Le système immunitaire se doit d’être étroitement régulé afin d’éviter que des
réponses immunologiques inappropriées ou de trop forte intensité ne surviennent. Ainsi,
différents mécanismes permettent de maintenir une tolérance périphérique, mais aussi
d’atténuer la réponse lorsque celle-ci n’est plus nécessaire. De tels mécanismes sont
cependant aussi exploités par les tumeurs, qui peuvent ainsi échapper à une attaque par le
système immunitaire et donc poursuivre leur progression. Ces mécanismes
immunosuppresseurs nuisent non seulement à la réponse naturelle contre les cellules
tumorales, mais font aussi obstacle aux tentatives de manipulation clinique de l’immunité
visant à générer une réponse anti-tumorale par l’immunothérapie.
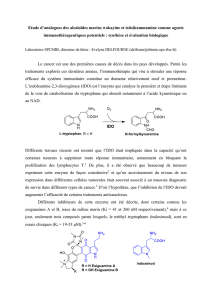
L’un des mécanismes par lesquels les tumeurs s’évadent du système immunitaire est
l’expression d’enzymes responsables du métabolisme des acides aminés dont l’une des
principales est l’indoleamine 2,3-dioxygénase (IDO). Cette dernière dégrade le tryptophane
et diminue ainsi sa disponibilité dans le microenvironnement tumoral, ce qui engendre des
effets négatifs sur la prolifération, les fonctions et la survie des lymphocytes T qui y sont
présents. Bien que la régulation de l’expression de cette enzyme ait été largement étudiée
chez certaines cellules présentatrices d’antigènes, dont les macrophages et les cellules
dendritiques, peu est encore connu sur sa régulation dans les cellules tumorales humaines.
Nous avons posé l’hypothèse que différents facteurs produits par les cellules
immunitaires infiltrant les tumeurs (TIIC) régulent l’expression de l’IDO dans les cellules
tumorales. Nous avons effectivement démontré qu’une expression de l’IDO est induite chez
les cellules tumorales humaines, suite à une interaction avec des TIIC. Cette induction
indépendante du contact cellulaire résulte principalement de l’interféron-gamma (IFN-γ)
produit par les lymphocytes T activés, mais est régulée à la baisse par l’interleukine (IL)-
13. De plus, la fludarabine utilisée comme agent chimiothérapeutique inhibe l’induction de
l’IDO chez les cellules tumorales en réponse aux lymphocytes T activés. Cette observation
pourrait avoir des conséquences importantes en clinique sachant qu’une forte proportion

ii
d’échantillons cliniques provenant de tumeurs humaines exprime l’IDO. Enfin, les
lymphocytes B, qui sont retrouvés également dans certaines tumeurs et qui interagissent
étroitement avec les lymphocytes T, sont aussi susceptibles à une induction
transcriptionnelle et traductionnelle de l’IDO. Cette enzyme est cependant produite sous
une forme inactive dans les lymphocytes B, ce qui rend peu probable l’utilisation de l’IDO
par les lymphocytes B comme mécanisme pour freiner la réponse immunitaire.
Nos travaux apportent des informations importantes quant à la régulation de
l’expression de la molécule immunosuppressive IDO dans les cellules cancéreuses. Ils
démontrent que l’expression de l’IDO est influencée par la nature des cytokines présentes
dans le microenvironnement tumoral. De plus son expression est inhibée par la fludarabine,
un agent utilisé pour le traitement de certains cancers. Ces données devraient être prises en
considération dans la planification de futurs essais immunothérapeutiques, et pourraient
avoir un impact sur les réponses cliniques anti-tumorales.

iii
Mots-clés
Immunothérapie
Cancer du sein
Cancer du rein
Humain
Indoleamine 2,3-dioxygénase
Lymphocytes T activés
Interféron-gamma
Interleukine-13
Lymphocytes B
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
1
/
235
100%