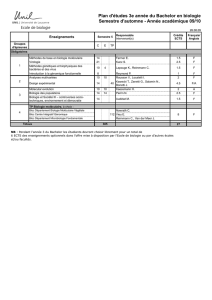
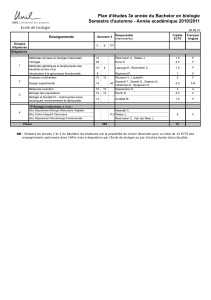
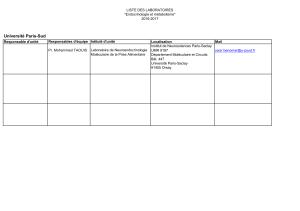
Dynamique moléculaire - Réseau Français de Chimie Théorique


Dynamique moléculaire
Outline
Introduction
Objectifs – méthode
Historique
Principe de la dynamique moléculaire
Séquence de mise en pratique
Comment obtenir le potentiel ?
Les formalismes
Formalisme de Newton
Formalisme de Lagrange
Formalisme de Hamilton
Les algorithmes de propagation
Le propagateur de Verlet
Le propagateur Velocity-Verlet
Les dynamiques moléculaires ab initio
Dynamique de Ehrenfest
Dynamique Born-Oppenheimer
Dynamique moléculaire Car-Parrinello

Dynamique moléculaire
Introduction
Objectifs – méthode
Objectifs et méthode
1. Calculer des moyennes statistiques d’un système à l’équilibre
2. Idem Monte Carlo
3. Principe ergodique :
hA(r(t),p(t))itime =hA(r,p)iensemble (1)
4. Echantillonner l’espace des phases
5. Technique “temporelle" d’échantillonnage plutôt qu’aléatoire comme en
MC
6. Evolution dans le temps du système


 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
1
/
27
100%