L`insuffisance rénale terminale est une maladie inflammatoire

L’insuffisance rénale terminale est une maladie
inflammatoire chronique
Ph. Rieu
Service de néphrologie et CNRS FRE 2534, Centre hospitalier universitaire de Reims
éditorial
Néphrologie Vol. 24 n° 7 2003, pp. 329-333 329
Ces dix dernières années, avec la découverte des récepteurs
« Toll », d’importants progrès ont été accomplis dans la compré-
hension des mécanismes moléculaires impliqués dans la réponse
immunitaire innée. La première partie de cette revue s’intéresse
à définir la réaction inflammatoire en intégrant ces nouvelles
données. La seconde partie de cet article est consacrée à l’in-
flammation au cours de l’insuffisance rénale terminale. Elle sert
d’introduction à ce numéro spécial de Néphrologie: « Inflamma-
tion en dialyse, une préoccupation quotidienne ». A travers ce
numéro, nous espérons vous convaincre que l’insuffisance rénale
terminale est une maladie inflammatoire chronique et que la
connaissance des causes et des conséquences impliquées dans
l’inflammation des malades dialysés permettra d’améliorer leur
prise en charge thérapeutique.
■La réaction inflammatoire
●
La réaction inflammatoire : une réponse immunitaire
innée contre les micro-organismes pathogènes
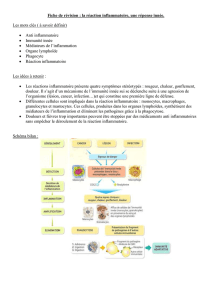
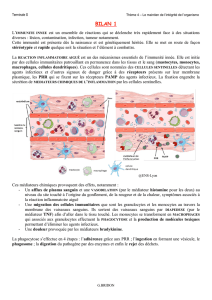
Pour se défendre contre les micro-organismes, l’homme dis-
pose de l’immunité innée et de l’immunité adaptative (tableau I).
L’immunité innée est un système de défense ancestral commun à
tous les êtres multicellulaires.1-3 Elle permet de neutraliser très rapi-
dement tout agent agresseur, puis d’orienter dans un deuxième
temps la réponse immunitaire adaptative. Les armes de ce système
de défense naturelle sont les phagocytes (macrophages et polynu-
cléaires), les peptides antimicrobiens et le complément. Contraire-
ment à l’immunité adaptative, les moyens de reconnaissance utili-
sés par ce système de défense préexistent à l’invasion par les
micro-organismes. Les signaux dangers reconnus par l’immunité
Résumé • Summary
Ces dix dernières années, avec la découverte des récepteurs
« Toll », d’importants progrès ont été accomplis dans la compré-
hension des mécanismes moléculaires impliqués dans la réponse
immunitaire innée. La première partie de cette revue s’intéresse
à définir la réaction inflammatoire en intégrant ces nouvelles
données. La seconde partie de cet article est consacrée à l’in-
flammation au cours de l’insuffisance rénale terminale. Elle sert
d’introduction à ce numéro spécial de Néphrologie : « Inflamma-
tion en dialyse, une préoccupation quotidienne ». A travers ce
numéro, nous espérons vous convaincre que l’insuffisance rénale
terminale est une maladie inflammatoire chronique et que la
connaissance des causes et des conséquences impliquées dans
l’inflammation des malades dialysés permettra d’améliorer leur
prise en charge thérapeutique.
Mots-clés: Inflammation – Insuffisance rénale terminale.
For the past ten years, the discovery of the Toll-like receptor
family have greatly contributed to our current understanding
of the innate immune system. The first part of this article draws
up the inflammatory reaction by integrating these new data. The
second part deals with inflammation during end stage renal
disease and introduces this special edition of Nephrologie :
« Inflammation in dialysis, a daily concern ». Through this edi-
tion, we would like to convince you that end stage renal failure is
a chronic inflammatory disease and that better understanding of
causes and consequences implied in dialysed patients inflamma-
tion will enable to improve their global therapeutic care.
Key words: Inflammation – End stage renal disease.
Tableau I : L’immunité innée et l’immunité adaptative.
Immunité Immunité adaptative
Etres multicellulaires Phylogénie Vertébrés
En quelques minutes Réponse En quelques jours
Phagocytes Lymphocytes
Peptides antimicrobiens Armes et immunoglobines
Complément
Antigènes conservés Signal danger Antigènes du non-soi
des agents pathogènes
(PAMP)
~ 100 PRR Récepteurs 1014 immunoglobulines
1018 TCR
Germinale Transmission Somatique

innée sont des déterminants moléculaires conservés des agents
pathogènes tels que les lipopolysaccharides (LPS), les peptidogly-
canes des parois bactériennes, l’ADN non méthylé des bactéries,
l’ARN double brins des virus. Ces antigènes conservés des agents
pathogènes ont été appelés PAMP (Pathogen associated molecu-
lar pattern) par C. Janeway (fig. 1). Les PAMP sont reconnus par
des récepteurs appartenant à l’immunité innée et appelés PPR
(pattern recognition receptors) (tableau II). Les PPR sont des
récepteurs sécrétés (C reactive protein, manose binding lectin),
membranaires (récepteurs Toll, macrophage scavenger receptor)
ou intracellulaires (protein kinase PKR). La réponse immunitaire
innée, ou réaction inflammatoire, résulte de l’interaction des PPR
avec les PAMP. Par exemple, la liaison de la CRP (C-reactive pro-
tein) avec les phosphocholines des parois bactériennes conduit:
1) à l’activation de la voie classique du complément qui attire les
cellules phagocytaires puis 2) à la phagocytose de ces bactéries
opsonisées par les cellules inflammatoires dont les récepteurs
Fc des immunoglobulines (FcγR) interagissent avec la CRP.4,5 La
famille Toll est une famille de PPR membranaires qui joue un rôle
central dans la réponse immunitaire innée. Le récepteur Toll a été
initialement décrit comme une protéine essentielle à l’établisse-
ment de l’axe dorso-ventral de la drosophile au cours de l’em-
bryogenèse. Il a été ensuite impliqué dans la réponse antifon-
gique de la drosophile. La voie de transduction qui conduit le
signal de Toll est structuralement similaire à la voie d’activation
de NF-κB à partir du récepteur de l’IL-1 au cours de la réponse
immunitaire chez les mammifères. En 1997, un homologue
humain de Toll (Toll like receptor 4 TLR4) a été décrit par l’équipe
de C. Janeway. Depuis, six récepteurs Toll ont été rapportés chez
les mammifères.1-3 Ces récepteurs sont exprimés par les cellules
phagocytaires, les cellules dendritiques, et certaines cellules épi-
théliales dont les cellules endothéliales. Leurs voies de signalisa-
tion sont conservées et font intervenir le facteur de transcription
NF-κB (fig. 2). Il semble que les différents membres de la famille
Toll soient impliqués dans la détection de PAMP spécifiques des
diverses classes de micro-organismes (fig. 3). TLR4 est nécessaire
à l’activation cellulaire induite par le LPS. TLR2 est impliqué dans
l’activation induite par les peptidoglycannes. L’interaction des
récepteurs Toll avec les PAMP conduit: 1) à la libération d’agents
anti-microbiens (dérivés réactifs de l’oxygène, enzymes protéoly-
tiques et peptides antimicrobiens) qui vont détruire l’agent
pathogène mais qui peuvent aussi avoir des effets « collatéraux »
sur les tissus; 2) à la production de cytokines et de chimiokines
qui avertissent et préparent l’organisme au danger et 3) à l’ex-
pression membranaire des molécules de costimulation (B7…) qui
vont initier la réponse immunitaire adaptative.1-3
●
La réaction inflammatoire : une réponse immunitaire
innée contre certains antigènes du soi
Les récepteurs PPR peuvent aussi reconnaître des antigènes
du soi. La CRP se lie aux phosphocholines exposées sur le feuillet
externe de la membrane plasmique des cellules en train de mou-
rir.4La protéine HSP60 (Heat shock protein 60) libérée par les cel-
éditorial
Néphrologie Vol. 24 n° 7 2003
330
Bactéries
Parasites
Levures
Virus
LPS
Peptidoglycan
Acide lipoteichoic
Lipoproteins
Lipoarabinomannan
Mannans
N-formyl-Met
ADN non méthylé
ARN double brins
Fla
g
ellin
Fig. 1: Identification de l’agent pathogène par les PAMP (Pathogen
associated molecular patterns).
Tableau II : Identification de l’agent pathogène par les PPR (pattern
recognition receptors).
Sécrétés Membranaires Intracellulaires
•C-Reactive Protein
(CRP)
•Serum Amyloid
Protein (SAP)
•Mannose binding
lectin
•Macrophage
mannose receptor
•
Macrophage
scavenger receptor
•TOLL-R
•PKR (ds RNA –
activated Protein
kinase
•OAS (Oligoadeny-
late synthase)
•NOD
α β
TLR4
CD14
LBP
LPS
IKK
I
Κ
B
NF
Κ
B
NIK TRAF6
IRAK
Transcription
MyD88
Fig. 2 : Voie de signalisation intracellulaire de TLR4.
LPS
Liptocheic acid
Peptidoglycan
Lipoprotein
Zymosan
Flagellin CpG DNA
TLR4 TLR1 TLR2
Monocytes/macrophages; polynucléaires; cellules dentritiques;
cellules endothéliales; cellules épithéliales intestinales
TLR6 TLR5 TLR9
Fig. 3 : Les récepteurs Toll des mammifères.

lules nécrosées se lie au récepteur TLR4.6Ces antigènes du soi
sont normalement inaccessibles aux récepteurs PPR. Libérés lors
de la nécrose cellulaire, ils pourraient déclencher la réaction
inflammatoire qui survient après une agression tissulaire quelle
que soit son origine (agression chimique, mécanique ou
hypoxique). Une modification biochimique des antigènes du soi
peut aussi induire l’apparition de déterminants moléculaires
reconnus par les récepteurs PPR. Par exemple, lorsque les LDL
(Low density lipoprotein) sont oxydés, ils se lient au récepteur
« scavenger » des macrophages qui est un récepteur PPR mem-
branaire.7Dans certaines circonstances, les antigènes du soi peu-
vent donc déclencher une réaction inflammatoire.
●Réaction inflammatoire et susceptibilité génétique
de la réponse
Certains récepteurs PPR présentent des polymorphismes
génétiques qui s’associent à des effets fonctionnels. Par exemple,
le polymorphisme Asp299/Gly de TLR4 diminue la réponse inflam-
matoire au LPS.8Ce polymorphisme est présent sous forme hété-
rozygote chez environ 6,5% de la population aux Etats-Unis. Il est
intéressant de noter que les taux sériques des marqueurs systé-
miques de l’inflammation (CRP, interleukine-6, procalcitonine,
néoptérine, fibrinogène, etc.) chez ces sujets sont plus bas que
chez les individus ne présentant pas ce polymorphisme.9Cet
exemple montre que les polymorphismes des récepteurs PPR par-
ticipent à la susceptibilité génétique gouvernant l’intensité de la
réaction inflammatoire.
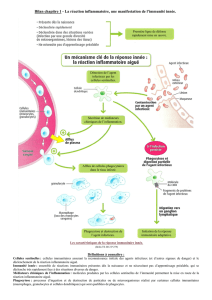
La réaction inflammatoire résulte donc de l’activation des cel-
lules impliquées dans l’immunité innée. Cette activation est
secondaire à la stimulation des récepteurs PPR par un « signal
danger ». Les signaux dangers sont des déterminants molécu-
laires appartenant aux micro-organismes pathogènes (PAMP) ou
à certains antigènes du soi. L’organisme est en permanence
confronté aux micro-organismes et à la mort cellulaire. Il est
donc très probable que de multiples réactions inflammatoires
infra-cliniques se déroulent en permanence dans l’organisme. La
sévérité de la réaction inflammatoire est liée à l’intensité du
« signal danger » et à la susceptibilité génétique de l’individu.
■Dialyse et inflammation chronique
●Les marqueurs de l’inflammation
L’intensité d’une réaction inflammatoire systémique peut être
mesurée avec l’aide de marqueurs sériques.10 Ces marqueurs
peuvent être des cytokines pro-inflammatoires produites par les
phagocytes activés, des protéines de la phase aiguë synthétisées
par les hépatocytes sous l’effet des cytokines, ou des protéines
d’adhérence solubles libérées par les cellules endothéliales acti-
vées. Le marqueur le plus utilisé en pratique clinique et lors des
études épidémiologiques est la CRP. Le rôle biologique et l’intérêt
clinique de cette protéine parmi les autres marqueurs de l’inflam-
mation sont détaillés dans les articles de Jean-Paul Cristol et
Franck le Roy.
Toutes les études épidémiologiques transversales réalisées
chez les sujets dialysés montrent que les marqueurs de l’inflam-
mation sont augmentés chez une proportion élevée de malades
(article de Christian Combe). L’intensité de cet état inflamma-
toire varie avec le temps chez chaque individu. Cependant, les
études épidémiologiques longitudinales dont l’étude BICHE rap-
portée par Christian Combe montrent clairement que l’inflam-
mation chronique, malgré ses variations, reste globalement
stable dans le temps. L’insuffisance rénale chronique terminale
est donc une maladie inflammatoire chronique.11,12
●Les causes de l’inflammation en dialyse
Les causes de cet état inflammatoire chronique sont mul-
tiples et font l’objet de plusieurs chapitres dans ce numéro spé-
cial de Néphrologie.
La maladie rénale initiale (glomérulonéphrite, pyélonéphrite
chronique, etc.) peut déjà être une source d’inflammation. La
diminution du catabolisme rénal des cytokines pro-inflamma-
toires parallèle à la diminution du débit de filtration glomérulaire,
en amplifiant les réponses cytokiniques, pourrait accroître l’inten-
sité des réactions inflammatoires. L’état urémique, en accélérant
la transformation des antigènes du soi par oxydation, glycoxyda-
tion ou carbamylation, pourrait induire l’interaction de ces anti-
gènes modifiés aux récepteurs PPR et activer ainsi la réponse
immunitaire innée. Les AOPP (Advanced oxidation protein pro-
duct) pourraient être un exemple de ce phénomène (article de
Béatrice Descamps-Latscha). Les AOPP résultent de l’oxydation
chlorée des protéines. Leur taux sérique augmente considérable-
ment au cours de l’insuffisance rénale chronique. Ces AOPP sti-
mulent in vitro l’explosion respiratoire et la production de cyto-
kines pro-inflammatoires par les phagocytes. Ils pourraient ainsi
participer à l’inflammation. Leur récepteur à la surface des phago-
cytes est encore inconnu. Au contraire, d’autres toxines uré-
miques telles que le p-cresol inhibent la fonction des cellules de
l’immunité innée (article de Raymond Vanholder). L’immunodé-
pression engendrée par ces toxines pourrait favoriser la diffusion
systémique des micro-organismes et augmenter ainsi l’exposition
aux agents pathogènes. L’exposition aux germes est aussi facilitée
par les accès de dialyse qui sont une porte ouverte de l’organisme
vers le monde extérieur, et les « bio-films » des cathéters ou des
circuits extracorporels (article de Bernard Canaud). Enfin, malgré
les efforts des industriels, la bio-incompatibilité des matériaux et
des solutions de dialyse reste un facteur important contribuant à
l’inflammation chronique du dialysé (articles de Jacques Chanard
et de Jean-Philippe Ryckelynck).
●Les conséquences de l’inflammation en dialyse
Malnutrition, douleur, résistance à l’érythropoïétine, et athé-
rosclérose sont les principales conséquences de l’inflammation
chronique.
La réorientation des priorités énergétiques qui accompagne
une inflammation systémique peut conduire à un état de mal-
nutrition. Chez le malade dialysé, l’inflammation chronique est
un facteur important participant à la dénutrition (articles de
Denis Fouque et de Thierry Lobbedez).
La douleur fait partie des signes cardinaux de l’inflammation.
Elle pose des difficultés de prise en charge chez le malade dia-
lysé. Ce thème est traité dans les articles de Frédéric Guirimand
et de Louis Brasseur.
éditorial
Néphrologie Vol. 24 n° 7 2003 331

La résistance de l’hématopoïèse à l’érythropoïétine est une
conséquence de l’inflammation bien connue des néphrologues. La
découverte récente de l’hepcidine permet de mieux comprendre
ce phénomène. L’hepcidine est un peptide antimicrobien produit
par le foie au cours d’une réaction inflammatoire.13 Ce peptide est
capable de détruire certaines bactéries telles que Escherichia coli.
L’hepcidine est aussi une hormone qui a une action sur les cellules
cryptiques intestinales et sur les macrophages. Elle augmente l’en-
trée du fer dans ces cellules en agissant sur le récepteur à la trans-
ferrine-1 (Trf1). L’accumulation de fer dans le cytoplasme des cel-
lules cryptiques induit, lors de la maturation de ces cellules en
entérocytes, une diminution de l’expression apicale du transpor-
teur du fer DMT1. La production d’hepcidine au cours de l’inflam-
mation s’associe donc à un stockage du fer dans les macrophages
et à une diminution de l’absorption intestinale du fer. L’hepcidine
est donc une protéine de la phase aiguë de l’inflammation qui
induit une carence fonctionnelle en fer et par conséquent une
diminution de l’hématopoïèse.14-16
L’athérosclérose est une maladie inflammatoire chronique de
l’intima des artères de gros et moyen calibres (article d’Alain Ted-
gui). L’agent d’agression entraînant la réaction inflammatoire est
le cholestérol-LDL sous une forme oxydée. Les LDL oxydés s’ac-
cumulent dans l’intima, activent les cellules endothéliales et faci-
litent le recrutement des monocytes-macrophages circulants qui
endocytent en grande quantité les LDL oxydés et se transforment
en cellules spumeuses.17,18 Dans la population générale, mais
aussi chez le malade dialysé, plusieurs études prospectives ont
montré que des taux sériques modérément élevés des mar-
queurs de l’inflammation représentent un facteur de risque vas-
culaire indépendant des facteurs de risque traditionnels (article
d’Alain Tedgui). L’interprétation de ces données est complexe.
L’athérosclérose pourrait être la cause de l’inflammation systé-
mique. Dans cette hypothèse, l’augmentation des marqueurs de
l’inflammation ne serait que la traduction systémique des mul-
tiples réactions inflammatoires intimales. L’inflammation systé-
mique ne ferait alors que refléter la sévérité de la maladie athéro-
scléreuse. Au contraire, l’athérosclérose pourrait être, non pas la
cause, mais la conséquence de l’inflammation systémique. Dans
cette hypothèse, une inflammation systémique quelle que soit
son origine, en modifiant le climat cytokinique et en activant
ainsi de façon aspécifique les cellules inflammatoires présentes
dans l’intima, pourrait favoriser la progression des lésions d’athé-
rosclérose. Plusieurs exemples dans la littérature vont dans ce
sens. L’injection intra-péritonéale d’une cytokine pro-inflamma-
toire telle que l’IL-6 accélère les lésions d’athérosclérose des sou-
ris sous régime athérogène.19 Les injections répétées de lipopoly-
saccharides bactériens chez les lapins sous régime athérogène
aggravent les lésions athéroscléreuses.20 Chez l’homme, les
infections bactériennes chroniques représentent un facteur de
risque d’artériosclérose.21 Enfin, le polymorphisme faible répon-
deur de TLR4 (Asp299/Gly) diminue le risque d’artériosclérose.9
Ces données suggèrent que l’inflammation systémique pourrait
être un facteur de progression de la maladie athéromateuse.
Intervenir sur l’inflammation pourrait alors ralentir la progression
de l’athérome.
●Prise en charge de l’inflammation chez l’urémique
L’inflammation systémique des malades dialysés peut être
réduite en s’attaquant à ses causes. Dans ce but, il faut chercher
à intensifier l’asepsie des voies d’abord de dialyse, à limiter les
« biofilms », à accroître les performances de l’épuration extraré-
nale, et à améliorer la biocompatibilité des matériaux et des solu-
tions de dialyse. La connaissance des mécanismes moléculaires
impliqués dans l’activation de la réponse immunitaire innée au
cours de l’urémie permettra dans un futur proche d’identifier des
cibles thérapeutiques spécifiques. En attendant, les traitements
anti-inflammatoires classiques tels que les anti-inflammatoires
non stéroïdiens, les statines, et les anti-oxydants peuvent avoir
leur place (article de Ziad Massy). En limitant l’inflammation sys-
témique, ils pourraient réduire ses conséquences. Leurs prescrip-
tions dans ce sens devront attendre les résultats d’études cli-
niques réalisées chez le malade dialysé.
Adresse de correspondance:
Pr Philippe Rieu
Service de néphrologie
Centre hospitalier universitaire de Reims
Hôpital Maison Blanche
45, rue Cognacq-Jay
F-51092 Reims
E-mail: [email protected]
1. Medzhitov R, Janeway C. Innate Immunity. N Engl J med 2000; 343 :
338-44.
2. Aderem A, Ulevitch RJ. Tool-like receptors in the induction of the innate
immune response. Nature 2000; 406 : 782-7.
3. Janeway CA, Medzhitov R. Innate Immune Recognition. Annu Rev Immu-
nol 2002; 20 : 197-216.
4. Hack E, Wolbink GJ, Schalkwijk C, Speijer H, Hermens WT, Van den Bosch
H. A role for secretory phospholipase A2and C-reactive protein in the
removal of injured cells. Immunol Today 1997 ; 18: 111-5.
5. Stein MP, Mold C, Du Clos TW. C-Reactive Protein Binding to Murine Leu-
kocytes Requires Fcg Receptors. J Immunol 2000; 164 : 1514-20.
6. Ohashi K, Burkart V, Flohé S, Kolb H. Cutting Edge: Heat shock protein
60 Is a putative endogenous ligand of the toll-like receptor-4 complex.
J Immunol 2000; 164 : 558-61.
7. Steinberg D. Low density lipoprotein oxidation and its pathobiological
significance. J Biol Chem 1997; 272 : 20963-6.
8. Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Fress K,
Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin
hyporesponsiveness in humans. Nat Genet 2000; 25 : 187-91.
9. Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E,
Willeit J, Schwartz D. Tool-like receptor 4 polymorphisms and atheroge-
nesis. N Engl J Med 2002; 347 : 185-92.
10. Gabay C, Kushner I. Acute-phase proteins and other systemic responses
to inflammation. N Engl J Med 1999; 340 : 448-54.
11. Stevinkel P, Alvesrtand A. Inflammation in End-stage renal disease: Sources,
consequences, and therapy. Semin Dial 2002 ; 15 : 329-37.
éditorial
Néphrologie Vol. 24 n° 7 2003
332
Références

12. Kaysen GA. The microinflammatory state in uremia: Causes and poten-
tial consequences. J Am Soc Nephrol 2001; 12 : 1549-57.
13. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial
peptide synthesized in the liver. J Biol Chem 2000 ; 276: 7806-10.
14. Fleming RE, Sly WS. Hepcidin: A putative iron-regulatory hormone rele-
vant to hereditary hemochromatosis and the anemia of chronic disease.
PNAS 2001; 98 : 8160-2.
15. Nicolas GN, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A,
Vaulont S. Lack of hepcidin gene expression and severe tissue iron over-
load in upstream stimulatory factor 2 (USF2) knockout mice. PNAS 2001;
98: 8780-5.
16. Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B,
Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia
in transgenic mice expressing liver hepcidin. PNAS 2002; 99 : 4596-601.
17. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med 1999;
340: 115-26.
18. Glass CK, Witztum JL. Atherosclerosis: The road ahead. Cell 2001 ; 104 :
503-16.
19. Huber S, Sakkinen P, Conze D. Interleukin-6 exacerbates early atheroscle-
rosis in mice. Arteriosclerosis Thromb Vasc Biol 1999 ; 19: 2364-7.
20. Lehr H, Sagban T, Ihling C, Zähringer U, Hungerer K, Blumrich M, Reifen-
berg K, Bhakdi S. Immunopathogenesis of atherosclerosis: Endotoxin
accelerates atherosclerosis in rabbits of hypercholesterolemic diet. Circu-
lation 2001; 104 : 914-20.
21. Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F,
Muggeo M, Xu Q, Wick G, Poewe W, Willeit J. Chronic infections and the
risk of carotid atherosclerosis – prospective results from a large popula-
tion study. Circulation 2001; 103 : 1064-70.
éditorial
Néphrologie Vol. 24 n° 7 2003 333
1
/
5
100%