INFECTIONS ET IMMUNODÉPRESSION AU COURS DE L`URÉMIE

FLAMMARION
MÉDECINE
-
SCIENCES
—
ACTUALITÉS
NÉPHROLOGIQUES
2006
(www.medecine.flammarion.com)
INFECTIONS ET IMMUNODÉPRESSION
AU COURS DE L’URÉMIE
par
Ph. RIEU et F. TOURÉ*
Les complications infectieuses, en particulier bactériennes, sont une source
importante de morbidité chez les patients insuffisants rénaux. Dans une étude pros-
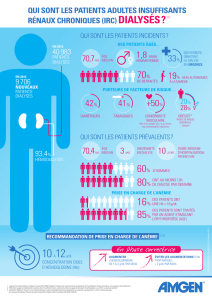
pective sur une cohorte de 1455 malades hémodialysés en France, la fréquence
des épisodes infectieux était de 39 pour 100 patients-année [1, 2]. Les principaux
syndromes infectieux étaient les pneumopathies, les infections de l’accès vascu-
laire et les infections urinaires (tableau I). Vingt-cinq pour cent des épisodes infec-
tieux étaient associés à une bactériémie. Dans cette étude, la fréquence des
pneumopathies était d’environ 13 épisodes pour 100 patients-année, alors qu’elle
n’est que de 1,5 épisodes pour 1000 sujets par an en France dans la population
générale. Dans l’étude HEMO menée aux États-Unis sur une cohorte de
1 846 malades hémodialysés, 42,4 p. 100 des patients étaient hospitalisés au moins
une fois en 34 mois pour une infection grave (tableau II) [3-5]. Les complications
infectieuses sont la deuxième cause de mortalité chez les patients insuffisants
* Service de Néphrologie –Transplantation, Hôpital Maison Blanche, et CNRS UMR 6198, Reims.
TABLEAU I. — FRÉQUENCES DES SYNDROMES INFECTIEUX DES MALADES HÉMODIALYSÉS.
INFECTIONS FRÉQUENCE (en p. 100)
Pneumopathie 34
Accès vasculaire 28
Infection urinaire 21
Parties molles/Os 10
Autres ou indéterminés 6

230 PH. RIEU ET F. TOURÉ
rénaux [6]. Elles sont à l’origine d’environ 20 p. 100 des décès des malades en
dialyse. En comparant avec la population générale et après ajustement pour l’âge,
le sexe et la présence d’un diabète, le risque de mourir d’un sepsis est multiplié
par vingt chez les patients transplantés et par cent chez les sujets dialysés [7] ; le
risque de mort par pneumopathie est multiplié par dix chez les malades dialysés
[8]. Cette augmentation du risque infectieux au cours de l’urémie est liée d’une
part à une exposition accrue aux agents infectieux et d’autre part à une anomalie
des défenses immunitaires [6, 9-13].
UNE EXPOSITION ACCRUE AU RISQUE INFECTIEUX
Rupture des barrières naturelles anti-infectieuses
L’organisme est confronté en permanence à un nombre gigantesque de micro-
organismes massivement présents aux portes d’entrées naturelles que sont la peau
et les muqueuses. La rupture de ces barrières naturelles, composantes de l’immu-
nité innée, augmente considérablement le risque d’infection.
BARRIÈRE CUTANÉE
Dans une étude rétrospective, sur presque 400 000 patients dialysés aux États-
Unis, le risque de septicémie était de 17,5 épisodes par 100 patients-année en
hémodialyse et de 8 épisodes par 100 patients-année en dialyse péritonéale [14].
Les staphylocoques étaient les principaux germes identifiés. Dans l’étude prospec-
tive multicentrique EPIBACDIAL menée par l’équipe de Nancy, les bactériémies
les plus fréquentes étaient aussi staphylococciques, avec très souvent pour point
de départ l’abord vasculaire [15]. L’incidence des bactériémies était de 11,16 épi-
sodes par 100 patients-année. Soixante-huit pour cent des germes isolés étaient
TABLEAU II. — FRÉQUENCE ET SÉVÉRITÉ DES SYNDROMES INFECTIEUX À L’ORIGINE D’UNE
HOSPITALISATION DES MALADES HÉMODIALYSÉS DANS L’ÉTUDE HEMO.
INFECTIONS FRÉQUENCE (en p. 100) DÉCÈS (en p. 100)
Endocardite 2,5 30,4
Infection urinaire 3,5 0
Parties molles/Os 3,5 9,1
Pied diabétique 4,5 10
Gastro-intestinale 6 21,2
Artérite 7,5 16,4
Pneumopathie 21 13,6
Accès vasculaire 22 6,6
Autres ou indéterminés 28,5 24

INFECTIONS ET IMMUNODÉPRESSION AU COURS DE L’URÉMIE 231
des staphylocoques. Les principaux facteurs de risque indépendants de bactériémie
étaient la présence d’un cathéter (RR = 7,6) et un antécédent d’au moins deux
épisodes de bactériémie (RR = 7,33). Ainsi, l’augmentation de la susceptibilité aux
infections bactériennes est en grande partie secondaire à la rupture de la barrière
cutanée. La peau saine est une barrière physique efficace contre la plupart des
agents microbiens qui ne peuvent normalement pas la franchir. Elle appartient à
l’immunité naturelle non spécifique. Chez les malades dialysés, cette première
ligne de défense est en permanence rompue par les cathéters veineux ou périto-
néaux, ou par les ponctions itératives de l’abord vasculaire.
BARRIÈRE UROTHÉLIALE
Toujours dans l’étude EPIBACDIAL, la proportion de bactériémie à bacille
Gram négatif était loin d’être négligeable avec un taux de 25 p. 100 (2,9 épisodes
par 100 patients-année). Les infections à germes à Gram négatif tirent leur origine
du tractus digestif ou uro-génital. Les infections urinaires ont été peu étudiées chez
les malades dialysés. L’incidence d’infection urinaire est pourtant élevée, de
l’ordre de 7,2 épisodes par 100 patients-année dans l’étude Lorraine [1]. Elles se
compliquent dans 22 p. 100 des cas de septicémie. Le risque d’infection à point
de départ urinaire est particulièrement élevé lorsque la diurèse résiduelle est très
faible, surtout lorsqu’il existe des antécédents d’infection du parenchyme rénal en
cas de lithiase ou de reins polykystiques. L’oligurie liée à l’insuffisance rénale
terminale et les modifications de la muqueuse vésicale qui en résultent, favorisent
la colonisation bactérienne. Ici encore, le risque infectieux est augmenté car la
barrière naturelle que représente la muqueuse urothéliale est endommagée.
Risque infectieux nosocomial
Les techniques invasives de dialyse, l’environnement hospitalier, et l’état uré-
mique favorisent la survenue d’infection nosocomiale. Les risques de contamina-
tion bactérienne et virale par les appareils de dialyse ou par le manuportage sont
bien connus. Le respect strict des mesures d’hygiène et des techniques de décon-
tamination des circuits extracorporels a permis de réduire considérablement ce
type de transmission infectieuse. Mais des facteurs autres que ceux liés à la dia-
lyse participent aussi au risque d’infection nosocomiale des sujets urémiques.
Dans une étude prospective menée à l’hôpital universitaire de Nashville (États-
Unis), le risque d’infection nosocomiale après ajustement sur la durée d’hospita-
lisation était plus élevé chez les malades dialysés (9,1/1 000 patients-jour) que
chez les sujets non dialysés (3,8/1 000 patients-jour, p < 0,001) [16]. Les co-
morbidités mesurées par le score de Charlson étaient supérieures chez les malades
dialysés et représentaient un facteur de risque d’infection nosocomiale. L’infec-
tion urinaire était l’infection nosocomiale la plus fréquente chez les sujets urémi-
ques (4,2/1 000 patients-jour), beaucoup plus fréquente que chez les sujets non
dialysés (0,7/1 000 patients-jour, p < 0,001). Le taux de sondage urinaire était
pourtant identique dans les deux groupes. Le germe le plus souvent impliqué dans
les infections urinaires nosocomiales était le Candida chez les malades dialysés
alors qu’il s’agissait d’E. coli chez les sujets non dialysés. Ces données montrent
bien qu’une conjonction de multiples facteurs (co-morbidités, sélection du
Candida par une antibiothérapie préalable, sondage d’une vessie urémique)
participe au risque élevé d’infections nosocomiales des malades dialysés. Les

232 PH. RIEU ET F. TOURÉ
soins médicaux chroniques, les hospitalisations itératives, l’utilisation fréquente
d’antibiotiques et les co-morbidités associées à l’état urémique (âge, diabète,
malnutrition) sont des facteurs qui participent, avec les techniques invasives de
dialyse, au risque élevé d’infections nosocomiales des malades dialysés.
UN DÉFICIT DE L’IMMUNITÉ INNÉE ET ADAPTATIVE
La susceptibilité des patients urémiques aux infections n’est pas expliquée seu-
lement par une exposition accrue au risque infectieux. Des troubles de l’immunité
participent à ce phénomène. Ils avaient été évoqués dès 1957 par Dammin devant
la survie prolongée des greffes de peau chez les sujets urémiques [17]. En effet,
les greffes de peau survivent 32 à 115 jours chez les patients urémiques, alors que
la survie n’est que de 8 à 10 jours pour les témoins. L’urémie s’accompagne d’ano-
malie de l’immunité innée et de l’immunité adaptative.
Immunité innée humorale
Le complément est le principal auxiliaire de l’immunité innée humorale. Le
complément comprend un ensemble d’environ 30 protéines plasmatiques syn-
thétisées essentiellement par le foie et les macrophages. Son activation en cas-
cade aboutit à la formation du complexe d’attaque membranaire, permettant la
formation d’un pore dans la membrane plasmatique des bactéries sensibles et
une lyse cellulaire osmotique. Les sous-unités C3a et C5a libérées ont en plus
un effet agoniste sur de nombreuses cellules, tout particulièrement sur les neutro-
philes. On distingue trois voies d’activation : 1) la voie alterne déclenchée par
la liaison du complexe C3bB à une surface activatrice (surface dépourvue de
glyco-aminoglycanes) ; 2) la voie classique activée par la liaison du C1q aux
complexes immuns ; et 3) la voie des lectines provoquée par la liaison de la MBP
(mannose binding protein) sur les sucres des parois bactériennes. Les dialyseurs
composés de membrane en cellulose (cuprophane) sont bien connus pour activer
le complément [18]. Cette activation est mise en évidence par la mesure de la
concentration de C5a, C3a ou de complexe C5b-9 plus élevée dans la ligne vei-
neuse que dans la ligne artérielle [19]. L’activation du complément semble faire
intervenir la voie alterne et la voie classique. La voie alterne pourrait être déclen-
chée par la liaison du C3b au groupement hydroxyle de la cellulose. La substitution
des groupes hydroxyles par l’acétate (acétate de cellulose), le diéthylamino-éthyl
(hémophane) ou un groupe benzyl (SMC) permet ainsi de diminuer l’intensité de
l’activation complémentaire [20]. La voie classique joue aussi un rôle car l’acti-
vation du complément par les membranes de dialyse en cellulose est retardée chez
les patients hypo-gammaglobulinémiques ou chez les patients avec un déficit géné-
tique complet en C4 [21]. Le sérum normal contient des anticorps anti-poly-
saccharidiques qui pourraient se fixer à la cellulose et déclencher l’activation de
la voie classique du complément [22]. Les membranes en cellulose modifiée et les
membranes synthétiques dites biocompatibles activent le complément à un degré
moindre. Par rapport au taux sérique basal, la concentration en C3a dans la ligne
veineuse est multipliée par 35 lors d’une dialyse avec un dialyseur en cuprophane,

INFECTIONS ET IMMUNODÉPRESSION AU COURS DE L’URÉMIE 233
par 10 avec un dialyseur en hémophane et par 5,7 avec un dialyseur en polysulfone
[19]. Les membranes en polyméthylmétacrylate, en polyacrylonitrile, en éthylène-
vinylalcool ou en polycarbonate activent aussi le complément in vivo [19, 23-30].
Les mécanismes d’activation du complément par les membranes synthétiques font
intervenir leurs propriétés d’adsorption protéique [31]. Ceci explique les capacités
variables des différents types de dialyseurs à activer le complément. Certaines
membranes ont la capacité d’adsorber les anaphylatoxines complémentaires (C3a
et C5a) libérées in situ [32]. Cette propriété pourrait limiter l’effet systémique de
ces médiateurs de l’inflammation. Sinon, la libération des anaphylatoxines com-
plémentaires induite par l’hémodialyse stimule les leucocytes et interfère ainsi
avec l’immunité innée cellulaire [33, 34]. Enfin, l’activation du complément par
l’hémodialyse pourrait inhiber son fonctionnement. En effet, l’activation in vitro
du complément après une séance de dialyse est altérée : la génération de C3c à
partir de sérum incubé in vitro avec du zymozan (voie alterne) ou des immuno-
globulines agrégées (voie classique) diminue après une séance de dialyse avec un
dialyseur en cellulose [35]. Cette inhibition de l’activation complémentaire persiste
plusieurs heures après la séance de dialyse. Elle est aussi constatée, mais avec un
effet moindre, après une séance de dialyse avec un dialyseur en cellulose modifiée
(polyéthylène glycol) ou composé d’une membrane synthétique (polyacryloni-
trile). Cette inhibition de l’immunité innée humorale mise en évidence in vitro
pourrait participer à la susceptibilité accrue aux infections bactériennes constatées
in vivo chez les patients urémiques.
Immunité innée cellulaire
L’immunité innée cellulaire désigne un ensemble de réponses non spécifiques et
identiques, que l’agression soit de nature physique ou microbienne. Ces réponses
sont rapides car elles ne nécessitent pas d’apprentissage ni d’expansion clonale et
constituent donc la première ligne de défense de l’organisme. Les principaux
acteurs en sont les polynucléaires, les macrophages et les cellules tueuses naturelles.
NEUTROPHILES
Le polynucléaire neutrophile assure une défense naturelle et ancestrale. Il est
un atout très important pour l’immunité innée, car il est le premier leucocyte à
migrer, en très grand nombre, vers le site infecté. Ses principales armes sont la
phagocytose, l’explosion respiratoire, la sécrétion de protéases et de facteurs cyto-
cides. Sa durée de vie est limitée, il meurt par apoptose en quelques heures. Le
rôle principal des neutrophiles est la clairance bactérienne [36, 37].
La fréquence élevée des infections bactériennes des patients urémiques telles
que les pneumopathies ou les parodontites, laisse penser qu’il existe un défaut de
fonction des neutrophiles [8, 38-41]. Cependant, il n’existe pas de test in vivo chez
l’homme pour démontrer le défaut de fonction de ces cellules. Très peu d’expé-
riences ont été rapportées chez l’animal. Nelson et al. ont trouvé que la migration
des neutrophiles dans les tissus infectés est retardée chez le rat urémique [42]. De
même, la clairance des bactéries injectées en intra-péritonéal est ralentie chez les
souris insuffisantes rénales [43]. Cependant, les différences observées in vivo sont
très faibles par rapport aux animaux contrôles et elles ne sont pas toujours mises
en évidence.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
1
/
22
100%