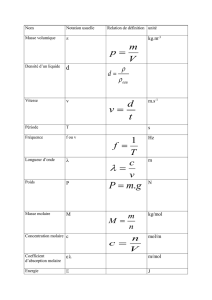
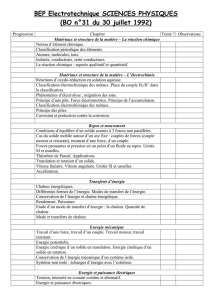
Dynamique Moléculaire en sciences des matériaux - mms2

Dynamique Moléculaire en
sciences des matériaux
Bernard Monasse
Ecole des Mines de Paris
CEMEF

Domaines d’application
Métaux, polymères, céramiques
Composition, structure chimique
Milieux denses, solutions, gazeux
Transformations hors d’équilibre
Conformations, cinétique, énergie

Echelles de description
L = 0,1nm – 0,3 nm L = 1 nm – 100 nm L = 100 nm – 1000 nm L = 1 µm – 100 µm
t = 10-13 st = 10-12 s -10-9 st = 10-6 s - 10-2 st = 0,1 s – 1 s
L = 0,1nm – 0,3 nm L = 0,5 nm – 2 nm L = 1 nm – 5 nm L = 20 nm – 500 nm
t = 10-14 st = 10-11 s -10-10 st = 10-8 s - 10-4 st = 0,1 s – 1 s
Métaux
Polymères

Surfaces
L = 0,1nm – 0,3 nm L = 1 nm – 5 nm L = 10 nm – 100 nm L = 1 µm – 100 µm
t = 10-14 s t = 10-12 s -10-10 s t = 10-8 s - 10-4 s t = 10-4 s – 10-2 s

Liens entre échelles
Mécanique quantique
Calcul électronique
Liaisons
Mécanique moléculaire
Calcul de mécanique newtonienne des
atomes
Phases cristallines
Mécanique méso
Mécanique d’éléments représentatifs
Grains et textures cristallines
Milieux continus :
Mécanique et thermique
Procédés, calculs de structure
femto seconde nano seconde milli seconde seconde temps
Distance
mètre
mm
100 nm
nm
loi de comportement
paramètres interaction
Champ de force
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
1
/
49
100%