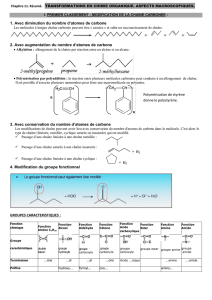
Algorithmes sur les séquences en bioinformatique

1
Algorithmes sur les séquences
en bioinformatique
Alessandra Carbone
Université Pierre et Marie Curie
M2 - STL
Cours 6 : Algorithmes d’alignement de structures
des protéines
A.Carbone - UPMC 2
Structure et représentation des protéines
Chaîne d’acides-aminés (1D)
Chaîne d’éléments structuraux
réguliers (2D)
Structure 3D d’une chaîne
d’acides-aminés.
Structure 3D de plusieurs chaînes
d’acides-aminés.
A.Carbone - UPMC 3
Les méthodes les plus communes pour trouver la structure 3D des protéines sont :
-Cristallographie – réalisée avec la diffraction des rayons-X et la diffraction des neutrons.
-Résonance Magnétique Nucléaire (Nuclear Magnetic Resonance - NMR)
Le fort intérêt pour des algorithmes de prédictions de la structure des protéines vient du fait
que ces méthodes sont lentes et chères.
La prédiction sera réalisée, quand cela est possible, à travers une comparaison 3D
de structures :
- la structure 3D est plus conservée à travers l’évolution que la séquence
- la structure 3D contient plus d’information que la séquence (e.g. sites actifs)
Problème de la comparaison structurale entre protéines
A.Carbone - UPMC 4
Quelques faits sur la structure 3D
Hypothèse de travail principale : une protéine se replie dans une structure unique,
qui est dépendante seulement de la séquence.
Un atome est modélisé par une balle ayant un rayon de van der Waals.
Le rayon de Van der Waals d’un atome est défini comme le rayon minimum qui ne permet
pas le chevauchement avec d’autres atomes.
Exemple de van der Waals radius

2
5
Chaque acide-aminé a un atome de carbone appelé Cα, qui est connecté à un groupe carboxyle
et un amino-groupe, un atome hydrogène et une partie qui dépend de l’acide amine spécifique,
appelé résidu.
Dans une chaîne protéique, l’amino-groupe d’un acide-aminé est connecté au carboxyle groupe
de l’acide-aminé qui le suit.
Cα
6
Les atomes Cαforment le squelette (backbone) de support de tous les autres atomes.
a. Cαde chaque acide-aminé
b. Tous les atomes formant le backbone du polypeptide
c. Liens hydrogènes qui permettent la formation des hélices α
a. b. c.
A.Carbone - UPMC 7
Méthodes pour la prédiction du repliement d’une protéine
Ils existent plusieurs approches pour approximer la structure d’une protéine :
Modélisation par homologie – utilise une base de données de protéines pour chercher des
séquences similaires. Si une protéines avec homologie d’au moins 30% est trouvée, c’est
assez probable que les deux protéines ont la même structure.
Threading – classifie structures connues dans des familles avec repliement similaire.
Étant donnée une séquence d’acides aminés, la méthode sélectionne la famille d’appartenance
la plus probable : aligne la protéine à une protéine similaire dont on connaît la structure,
et utilise la nouvelle structure comme point de départ pour effectuer des opérations
de repliement plus fines.
Ab initio – quand on a aucune information sur l’homologie d’une protéine on est forcé à replier
la protéine ab initio. Les solutions sont algorithmiquement difficiles même dans les modèles
les plus simples.
Exemple : si on divise les acides-aminés en hydrophiles et hydrophobes, on peut essayer de
construire la structure 3D en minimisant les répulsions et en maximisant les attractions entre
acides-aminés voisins. Il a été montré que la résolution de ce modèle est NP-complet, même
s’il y a des approches euristique avec ratio d’approximation 2/3.
A.Carbone - UPMC 8
Alignement structurale des protéines
Représentation 3D discrète de la forme d’une molécule. Caractéristiques critiques possibles :
- squelette des Cα
- squelette des Cαet Cβ
- éléments de la structure secondaire (hélices αet feuillets β)
- représentation de la surface moléculaire
Coordonnées Cαde deux molécules

3
A.Carbone - UPMC 9
Problème : Comparaison de deux structures
Entrée : un ensemble de coordonnées atomiques des deux molécules
Sortie : transformation rigide (rotation et translation) dans l’espace qui permet l’appariement
d’un nombre suffisant d’atomes d’une molécule aux atomes de l’autre.
Les algorithmes qui essaye de résoudre ce problème sont classifiables dans deux catégories:
1. Dépendant de l’ordre de la séquence. Utilise l’ordre des atomes dans la chaîne protéique,
en réduisant le problème à un problème de matching d’une courbe 3D (essentiellement un
problème 1D).
2. Indépendant de l’ordre de la séquence. Alignement de propriétés dans un espace 3D
(un problème 3D).
Avantages:
-Les biologistes sont parfois plus intéressés à des motifs qui préservent l’ordre de la séquence
-La puissance calculatoire nécessaire est réduite par l’usage de l’ordre d’une chaîne
Avantages:
- les techniques détectent des motifs 3D non-séquentiels, comme des motifs sur la surface de la
molécule, et en particulier les sites d’interaction.
- bdd structurelles sont approchables avec de l’information structurelle partielle
- l’algorithme peut être applicable à d’autres structures moléculaires
- l’algorithme n’est pas affecté par insertions et délétions.
A.Carbone - UPMC 10
On peux décomposer le problème de la comparaison structurale en deux :
1. matching – détection des propriétés de matching (difficile)
2. meilleure superposition par rapport à des propriétés de matching données – trouver une
transformation d’une structure dans l’autre avec RMSD (Root Mean Square Deviation:
elle mesure les différences entre les positions des atomes Cαentre deux protéines)
minimale.
Superposition (2) – le meilleur RMSD obtenu par alignement rigide
Entrée : deux ensembles de points en 3D : P = {pi} et Q = {qi} où i=1…n
Sortie : trouver une rotation 3D R0(cad retourner la figure autour d’un point a étant donné R0
degrés) et une translation a0(cad, chaque point de la figure bouge d’une distance a0dans
la même direction) telles que minR,a ∑i|Rpi+ a - qi|2 = ∑i|R0pi + a0-q
i|2
Solution : une solution pour ce problème existe et elle demande une analyse des eigenvalues
de la matrice de corrélation des points.
A.Carbone - UPMC 11
Un cas simple du problème de matching (1)
On considère le fait que les objets sont des corps rigides. Dans ce cas le matching d’une
paire de triplets de points ordonnés défini de façon unique une transformation 3D rigide.
Algorithme :
- Pour chaque paire de triplets, une pour
chaque molécule, définissant des triangles
« presque congruents », calculer la
transformation qui superpositionne les triangles.
- Calculer le nombre de paires de points qui sont presque superposées et donner un
score dépendant de ce nombre à la superposition des deux objets.
- Choisir la superposition qui obtient le score plus élevé et améliorer la transformation en
la remplaçant par la meilleur transformation RMSD, pour toutes paires matchées.
Complexité : O(n7), où O(n) est le nombre de points dans chaque molécule.
A.Carbone - UPMC 12
Algorithme d’alignement rigide basé sur les techniques d’hachage
géométrique (Wolfson et al. 1988, 2000)
Algorithme organisé en deux parties : preprocessing et reconnaissance
Preprocessing
Tous les modèles d’une bdd sont considérés. Pendant cette phase, pour chaque modèle,
son information géométrique est codifiée dans un tableau d’hachage.
Reconnaissance
Etant donné un objet, ses caractéristiques sont déduites. Ces caractéristiques sont utilisées
pour faire correspondre l’objets à plusieurs entrées dans le tableau d’hachage.
Une structure de référence 3D est un triplet
de vecteurs orthogonales ayant une origine
commune. Elle peut être définie de façon
unique par les sommets d’un triangle
non-dégénéré.
Exemple de structure
de référence 3D

4
A.Carbone - UPMC 13
Supposons ex, ey, ezles trois vecteurs unité. Chaque point en 3D peut être représenté
en utilisant la structure de référence comme v= αex+ βey+ γez+ p1.
La longueur des côtés du triangle que l’on veut stocker (CαNCβ) est invariante par translation
et par rotation, et en conséquence, on peut définir à partir de la structure de référence, une
signature de la forme en fixant les longueurs des côtés du triangle.
Algorithme :
Preprocessing. Pour chaque objet modèle fait ce qui suit::
1. Choisir une structure de référence.
2. Calculer la base orthonormale 3D associée à la structure de référence et sa signature
de forme (la longueur des côtés du triangle).
3. Calculer les coordonnées de tous les autres points (dans un voisinage pre-specifié) dans
cette structure de référence.
4. Utiliser chaque coordonnée comme une adresse du tableau d’hachage. Mémoriser
l’entrée (identificateur de protéine, structure de référence, signature de forme, points)
à l’adresse du tableau d’hachage.
5. Répéter les étapes 1-4 pour chaque structure de référence modèle (modèle de triplet
non-collinéaire de points).
A.Carbone - UPMC 14
Reconnaissance. Le matching de l’objet cible est réalisé comme suit :
1. Pour chaque structure de référence de l’objet cible :
a. Calculer la base orthonormale et la signature de la forme associée
b. Calculer les coordonnées de tous les autres points dans la structure de référence
courante.
c. Utiliser les coordonnées pour accéder au tableau de hachage et récupérer tous les
enregistrements (identificateur de protéine, structure de référence, signature de la
forme, points)
2. Pour les enregistrements avec une signature de la forme donnée, « voter » pour les
paires (identificateur de protéine, structure de référence).
3. Calculer les transformations des paires ayant des scores élevés. Pour chaque hypothèse,
il sera possible de calculer aussi les paires de points qui matchent. La liste des matching
et les transformations associées forment ce que l’on appelle « germe du matching ».
A.Carbone - UPMC 15
On peut maintenant construire un algorithme d’alignement qui utilise l’hachage géométrique
comme suit.
- définir le voisinage locale d’un résidu. Noter que si on utilise tous les trois points
pour chaque triplet, chaque atome sera représenté de façon redundante. Il apparaîtra dans la
table d’hachage dans toutes structures de référence. (En pratique, comme on n’est pas
intéressé aux atomes qui sont trop proches ou trop loin alors on prendra seulement les
atomes qui se trouvent dans un rayon entre un max et un min.)
- Appliquer la technique de l’hachage géométrique aux points voisins pour détecter les
germes du matching définis par une transformation et une liste de matching. Plusieurs listes
de matching pourrons partager la même transformation.
- On clustérise germes de matching et on réunie listes de matching
- Étape d’extension: les germes de matching sont étendus pour contenir des paires de
matching en plus.
- La meilleure transformation par rapport au RMSD est calculée. Pour cette raison, un
algorithme itératif heuristique qui minimise la somme des distances entre des nouvelles
paires est appliqué.
A.Carbone - UPMC 16
Complexité :
N nombre de protéines dans la bdd
O(n) nombre de caractéristiques par structure de protéine
R nombre de structures de référence, typiquement R= n, n2, n3
S taille de l’entrée du tableau d’hachage (s peut être maintenu petit)
Preprocessing O(N * R * n)
Reconnaissance O(R * N * s)

5
A.Carbone - UPMC 17
Algorithme d’alignement flexible basé sur les techniques d’hachage
géométrique (Wolfson et al. 2000)
Il y a deux types de mouvement possible pour une protéine : à charnières (hinge motion)
et de cisaillement (shear motion). L’approche d’hachage géométrique peut être étendu pour
traiter ces cas flexibles aussi. La méthode demande par contre la connaissance au préalable
de la position des charnières.
Entrée : deux molécules A = v1… vnet B = w1…wnreprésentées par leurs coordonnées Cα.
Sortie : décomposition des deux molécules dans un nombre minimal de fragments disjoints
de taille maximale, telle que le nombre de Cαdans chaque fragment est le plus proche
possible au nombre des Cαdu fragment matché et telle qu’il existe une translation rotationelle
en 3D qui superpose les atomes correspondants avec un plus petit RMSD.
18
1. Détecter des fragments rigides congruents qui soient suffisamment grands.
Construire une liste de paires d’atomes en correspondance (matching) constituée par
une seule paire tout au début. Itérativement, étendre la liste en ajoutant une paire
d’atomes à la gauche et une à la droite (en suivant la direction du backbone) jusqu’à
l’obtention de la plus longue paire de fragments congruents consécutifs qui inclue la
liste de paires d’atomes de départ. Pour calculer la liste de paires d’atomes
correspondants contigus, itérer cette étape pour tous les atomes Vi,Vj.
2. Trouver une séquence de fragments disjoints que suivra la séquence de Cαde A et de B.
La méthode est similaire à celle utilisée dans l’algorithme FastA:
a. Les listes de matching sont représentées comme
des nœuds d’un graphe.
a. Rejoindre deux nœuds par un arc, si la paire fragment
qu’ils représentent est consécutive dans l’alignement.
Les paires fragments peuvent chevaucher sur un
intervalle maximale de 2∆. Le résultat est un
graphe dirigé acyclique.
c. A chaque arc on assigne un poids de pénalité W(e).
L’idée étant de favoriser des fragments correspondants
très longs et de pénaliser les long gaps. Définir M1
et M2 deux listes d’atomes en correspondance
Vi…Vj avec Vk…Vt et Vb…Vf avec Vp…Vr.
où 0<i<j<f<n, b<f et 0<k<t<r<m, p<r
A.Carbone - UPMC 19
s’il n’y a pas de chevauchement, alors ∆= 0.
l est défini comme la longueur de M1.
gap1= b-j et gap2 = p-t
W(e)= - ( (l -1) - ∆ )2+ max ( |gap1|, |gap2|) + ||gap1|-|gap2||
M1M2f
r
i
k
j
t
b
p
gap1
gap2
d. Un nœud virtuel, appelé nœud de départ, et les arcs sortant de ce nœud et dirigés
vers tous les autres nœuds, ayant coût 0, sont ajoutés au graphe.
e. L’algorithme de recherche du chemin le plus court qui part du nœud de départ est
appliqué au graphe. Cela permettra le calcul du chemin le plus court à partir de chaque
nœud.
f. Tous les chemins possibles sont collectionnés et classés dans des groupes différents
par rapport au nombre des nœuds dans le chemin.
g. l’RMSD du chemin est calculé et les chemins de chaque groupe sont ordonnés par
rapport à cette valeur.
h. Pour chaque groupe, les 10 meilleurs résultats donnerons les possibles solutions,
chacune ayant un nombre de charnières différent.
Complexité de l’algorithme : O(k4), où k = max(|A|,|B|).
A.Carbone - UPMC 20
Représentation de la surface moléculaire
Une bonne représentation de la surface moléculaire aide à
-l’étude du repliement de la protéine,
-la prédiction de la reconnaissance biomoléculaire
-la détection des cavités jouant un rôle important dans l’interaction avec les médicaments.
Comme la complexité des algorithmes dépends du nombre de points représentants la surface
moléculaire, un problème important dans cette représentation concerne la dispersion des
points intéressants qui représentent la molécule.
 6
7
8
9
6
7
8
9
1
/
9
100%