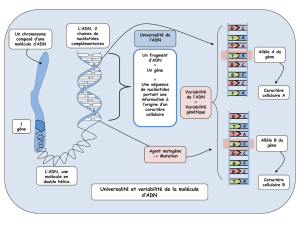
Biologie cellulaire du 13 janvier

1
Biologie cellulaire du 13 janvier.
Fondements de la biologie cellulaire.
Avant le 18ème siècle, on pensait qu’il n’y avait pas de points communs morphologique donc
fonctionnelle entre les êtres vivants, ce qui expliquait leurs diversité. Mais certain ont
considérés qu’il y avait peut être des briques communes entre ces êtres vivants. C’était qu’une
hypothèse jusqu’en 1665 où Hooke fait l’observation de logettes identiques nommées des
cellules. On a ensuite pu dire que tous les animaux contiennent des logettes ressemblant.
Schweden et Schwann en 1838 considèrent la cellule comme unité morphologique et
fonctionnelle commune aux êtres vivants. Il faut attendre Virchow pour démontrer qu’une
cellule provient toujours de la division d’une cellule antérieure, il complète la définition de la
cellule comme unité fondamentale de tout être vivant, c’est la plus petite portion de matière
vivante qui puisse vivre isolé et qui puisse se reproduire.
-Les êtres pluricellulaires ont créés la communication cellulaire afin que la fonction de chaque
cellule ou de toutes les cellules soit coordonnée et dès que la coordination disparaît c’est une
situation pathologique.
Toutes ces unités communes aux êtres vivants ont une unité d’organisation c’est à dire noyau,
organites identiques d’une cellule à l’autre, une unité de fonction avec un métabolisme
identique d’une cellule à l’autre et une unité de composition puisque les macromolécules
contenus dans ces cellules sont formés de petites molécules qui sont communes. On est donc
passé à une grande analogie entre les êtres.
- La prolifération cellulaire, la mort cellulaire sont des phénomènes programmés, régulés.
- Les mécanismes moléculaires qui sous tendent la fonction d’une cellule.
- Les interactions cellulaires qui coordonnent l’ensemble des fonctions ; exemple de la
transduction du signal qui fait passé un message de l’extérieur vers l’intérieur de la cellule.
- On utilisera les cellules du tissu nerveux et les cellules souches qui ont un intérêt
thérapeutique essentiel donc important de comprendre les régulations dans les cellules
souches, savoir comment on peut induire une différentiation vers un type cellulaire précis.
Comment une cellule est a l’origine d’une pathologie ? Ca peut être comme pour la
Mucoviscidose avec une mutation ponctuelle dans un gène. Le cancer est une pathologie
multifactorielle pour laquelle on peut définir des centaines d’altérations toutes responsables de
la pathologie.
- Quand on a identifié une étape qui ne marche pas bien, on peut imaginé des mécanismes
pour réparer cette étape : c’est l’utilisation de la biologie cellulaire en thérapeutique donc on
répare soit un gène, soit une cellule, soit on greffe une cellule.
Protocole de recherche d’une cible qui sera soit diagnostique soit thérapeutique.
On identifie un gène qui est sous exprimé ou sus exprimé dans une situation pathologique, on
va ensuite regardé si dans une cellule en culture on peut avoir un modèle de cette pathologie à
une échelle restreinte. Une fois cette pathologie trouvée, on va essayer de changer le
phénotype de cette cellule, la réparer.
- Exemple d’une cellule : On récupère des cellules qui viennent des patients et on les mets en
culture donc si on veut comprendre la pathologie de ce patient en particulier on le peut.
- Exemple de la manipulation génique : on fait exprimé un gène dans une zone du cerveau
chez le rat ou souris. On peut faire des transfères de gènes, greffer des cellules, pour cela on a
des souris transgéniques qui sont des modèles de pathologie (exemple de la souris
mucoviscidose).

2
Méthode descriptive :
Une cellule animale fait 10-20 micro mètre, c’est 5 fois plus petit que ce qu’on peut voir à
l’œil nu donc il a fallu attendre le microscope pour observer les cellules. Au microscope
photonique on peut observer des choses de 0,2 micromètres donc on peut voir la cellule et ses
structures grossières. Depuis les années 40 on a inventé le microscope électronique à balayage
ou transmission avec lequel on peut visualiser des objets de 1 nanomètre donc on peut
visualiser les macromolécules dans la cellule et définir précisément quelle est la morphologie
d’une cellule. Toute cellule à cette taille est translucide donc il a fallu à chaque fois des
colorants, des agents de contrastes pour mettre en valeur les structures et les connaissances
qui sont tirés de la microscopie doivent autant à la coloration qu’au microscope lui même.
Démarche de l’observation d’un tissu :
- La fixation du tissu : On utilise des aldéhydes comme le formaldéhyde (formol) qui font
des pontages entre les groupements aminés des protéines et toutes les protéines vont se
retrouvés piégés, réticulés, ça tue la cellule puisque ça fixe chaque molécule.
- Inclusion du tissu dans une matrice ou congelé pour les durcir car tissus trop mou pour être
coupés.
- On utilise des microtomes ou ultra microtomes (pour le ME) pour faire des coupes. On
utilise de la paraffine résine molle pour rigidifier assez le tissu pour le couper à une taille de
10 micromètre pour le microtome. Pour l’ultramicrotome on utilise l’époxy qui est une résine
très dure pour faire des coupes de quelques nanomètres.
- Tout cela est observable parce que coloré, contraster : L’hématoxyline est un colorant
nucléaire basique colorant les structures acides comme l’ADN, l’ARN. L’éosine colore les
éléments du cytoplasme. Il y a aussi le safran qui colore le collagène.
On a développé des techniques d’immuno histochimie (idem à l’immunofluorescence sauf
que le révélateur est différent) où un anticorps va reconnaître un antigène. L’exemple c’est la
mise en évidence du facteur de Von Vibrant ou facteur 8 de la coagulation qui est exprimé par
les cellules endothéliales ce qui va permettre de visualiser les vaisseaux. On peut mettre en
évidence PCNA c’est à dire un antigène nucléaire de prolifération cellulaire qui va permettre
de visualiser les cellules qui sont en division ; c’est intéressant parce que l’agressivité d’une
tumeur est corrélé à sa prolifération. GFAP est une protéine spécifique des astrocytes.
- Hybridation in Situ : faire la séquence complémentaire de ce que l’on recherche pour avoir
un sonde que l’on marque.
Toutes ces techniques ont une base commune : quelque soit la nature de la cible (protéine,
séquence nucléotidique, glucide, lipides) on définit une sonde qui est une séquence
complémentaire et on va ajouter un secondaire = un élément qui vient au dessus pour repérer
la sonde et permettre de visualiser la sonde car le secondaire contient une enzyme ou un
fluorofore repérable ensuite. On ne marque pas directement la sonde pour avoir un seul
secondaire pour toutes les techniques donc c’est économique. Il peut y avoir plusieurs
fluorochromes : des jaunes, des rouges pour faire plusieurs colorations à la fois, utilisation
aussi d’éléments radioactifs.
Culture cellulaire :
- Première expérience de culture cellulaire en 1907 pour régler une controverse en
neurologie : On ne savait pas si le prolongement des neurones venait de la cellule ou si c’était
la fusion de plusieurs cellules qui donnait ce prolongement. Pour cela des chercheurs ont mis
des échantillons de cerveau en culture dans un tampon à 37° / un liquide et ils ont regardés ce
qu’il se passait. Ils ont observés que de long prolongements apparaissaient au niveau de

3
chaque cellule mais pas observés de fusions de cellule. La conclusion c’est que le
prolongement venait d’une cellule, c’était l’hypothèse neuronale qui avait été vérifié.
Pendant longtemps on a pas sur faire de culture cellulaire mais on mettait un fragment de
tissus dans une condition espérée compatible avec sa survie. Il a fallu attendre de savoir
dissocier les cellules pour faire de la culture cellulaire. Dans un tissu les cellules sont
cohésives grâce à des molécules d’adhérences, elles adhèrent entre elles, à la matrice
extracellulaire et si on veut isoler les cellules il faut tout rompre.
D’abord on coupe au scalpel le tissu, ensuite on utilise des techniques enzymatiques qui
digèrent les éléments de la matrice extra cellulaire ou les molécules d’adhérences donc retirer
la cohésion du tissu. La trypsine dégrade un peu toutes les protéines. La collagènase est une
enzyme qui dégrade le collagène spécifique. Les molécules d’adhérences ont une fonction
dépendante du calcium donc si on piège le calcium ces molécules d’adhérences ne
fonctionnent plus et les cellules se dissocient, c’est le rôle de l’EDTA qui est un chélateur, un
piégeur du calcium. En général on utilise l’EDTA et une enzyme pour dissocier rapidement
un tissu.
On a donc ensuite un gros mélange de cellules qu’on peut mettre en culture et on peut faire un
trie des cellules à mettre en culture pour privilégier un type cellulaire. Pour cela on utilise des
techniques physico-chimiques : par exemple on sait que les macrophages adhérent facilement
au verre donc si on récupère ce qui n’a pas adhérer à une plaque de verre on a une population
déplétée en macrophage.
Si on connaît un antigène à la surface de la cellule que l’on veut récupérer, on marque la
cellule avec l’anticorps utilisé et il y a des moyens d’aller récupérer l’anticorps et la cellule
avec. Donc on peut trier toutes cellules pourvues qu’on en connaisse au moins un antigène
spécifique à la surface.
Définition sur la culture cellulaire.
Si on met les cellules isolées en culture, on a une culture primaire dérivant directement du
tissu. La culture primaire est polymorphe généralement, les cellules qui la compose
conservent les caractères de différentiation des cellules d’origines. Si on met ces cellules dans
une deuxième boite, on perds des cellules qui aiment pas la culture et surtout les cellules vont
commencer à se dédifférencier petit à petit. C’est à dire que les cellules épithéliales
intestinales vont perdre leurs morphologies spécifique de l’absorption dès le deuxième
passage. A cette étape on peut récupérer une seule cellule, la mettre en culture et la population
qui va naître dérivera d’une seule cellule par division successive donc cette population est la
plus homogène possible, a la même morphologie, la même fonction, le même patrimoine
génétique, c’est un clonage.
- Le microscope à contraste de phase est généralement utilisé.
- On peut faire de la vidéo microscopie pour suivre le déplacement, la division d’une cellule.
- En fluorescence on peut visualiser les flux de calcium en fonction de ce que l’on ajoute dans
la boite de culture donc on voit la cellule vivre réellement.
- Observation contraste interférentielle : Microscope qui permet de mettre en valeur toutes
les interfaces physiques et chaque membrane est une interface entre deux milieux donc on va
essentiellement voir les membranes cellulaires et intracellulaires avec ce microscope. Donc si
on utilise la sécrétion des cellules c’est intéressant.
- Ces mêmes cellules peuvent être observées en contraste de phase : on peut la cellule entière
avec le noyau, les nucléoles dans les noyaux, les étapes de la division, toutes cellules
arrondies est en train de se diviser et on voit une plaque métaphasique. Ces 2 petites cellules
très arrondies viennent de se diviser, viennent d’effectuer la cytodiérèse et on peut suivre
quelques organites.

4
Conditions de la culture :
-Il faut respecter les conditions physico-chimiques des cellules, une cellule doit rester à 37°, à
PH neutre, la force ionique doit être respecter, c’est la définition d’une solution saline
tamponnée. C’est ce qu’on utilise comme base de tous les milieux de culture (90%).
-On rajoute ensuite un élément qui contient des AA essentiels , des glucides, des vitamines ce
qui permet à la cellule de vivre et non de survivre. Des acides aminés non essentiel sont
produits par certains tissus comme la glutamine par le foie et le rein. C’est un milieu bien
définit, c’est son nom « milieu définit » mais tout cela n’est pas assez riche pour faire vivre
une cellule.
-Il lui faut des facteurs de croissance, des protéines et ceci est apporté par du sérum (de Veau)
à 10% du volume du milieu.
-Aujourd’hui on ne met pas de sérum pour réimplanter dans les patients donc on définit les
milieux donc on rajoute les protéines synthétisés in vitro, purifiés. 2 protéines indispensables
à rajouter dans un milieu de culture si on ne met pas de sérum, c’est l’insuline et la
transférine.
- Des cellules ne poussent pas facilement donc on leur met par exemple pour les cellules
endothéliales il faut mettre le VEGF (vascular endothelium growth factor) qui permet la
survie des cellules et stimule leur prolifération et il garantit leur différentiation (cellule
endothéliale reste cellule endothéliale). Il y a le NGF (neuronale) pour les cellules neuronales.
Il y a l’IL2 (interleukine 2) pour les lymphocytes et l’érythropoietine pour les cellules
dérivées des érythroblastes.
- Le support, le soutient des cellules : La plupart des cellules ne peuvent pas se permettre de
ne pas être adhérente (sauf les cellules du sang) donc la culture se fait sur du plastique pour
nombreuses cellules (comme macrophage) mais d’autres ont besoins d’un support particulier :
pour les cellules endothéliales micro vasculaires (capillaires) il faut reproduire un peu la
matrice du capillaire qui est la lame basale composée majoritairement de collagène 4 et de
fibronectine.
- Ici ce sont des mélanocytes tumoraux à gauche et à droite. A gauche on voit des
mélanocytes sans prolongements, sans mélanine, c’est la définition d’une cellule tumorale qui
se dédifférencie mais qui prolifère beaucoup. Sur ces mélanocytes on rajoute certains agents
et on regarde si ceux ci peuvent re différencier les mélanocytes et ici c’est le cas car il y a de
la mélanine dans les prolongements hors la différenciation est un des mécanismes recherchés
pour freiner la progression tumorale puisque quand une cellule se différencie elle ne prolifère
pas.
- Les cellules ne peuvent se diviser qu’en nombre réduit c’est à dire qu’on a peut de temps
pour utiliser la culture car les cellules meurent rapidement donc c’est pas très pratique.
-Donc on utilise souvent des cellules dites « immortalisées » c’est à dire qui ont perdus ce
contrôle qui les font mourir au bout d’un nombre finit de division. Pour obtenir des cellules
immortelles, certains virus comme SV40 (virus siliens) exprime dans la cellule une protéine
qui va dérégulé le contrôle de la prolifération cellulaire donc la cellule va proliférer de façon
permanente et pour toujours. On peut également déréguler l’expression de MYC qui est une
protéine cellulaire et on va rendre également les cellules immortelles.
- Il y a les cellules tumorales qui sont dites « transformées » qui sont immortelles mais
portent de nombreuses autres altérations (et pas uniquement sur le cycle cellulaire) qui font
que si cette cellule tumorale est réinjecter elle va formé une tumeur alors qu’une simple
cellule immortalisée ne le fera pas.
- La cellule LA : dérivé tumorale très étudié. C’est une cellule dérivé d’une tumeur du col
utérin, c’est une lignée mise en culture dans tous les labos du monde ce qui permet d’avoir le
même matériel pour faire certaines expériences et on peut comparer les résultats entre
laboratoires.

5
Différences entre cellules normales, immortelles et tumorales :
Plusieurs paramètres permettent de les séparer : le nombre de division, l’ancrage (a t’elle
besoin être ancré pour survivre ?), la dépendance aux facteurs de croissance, l’inhibition de
contact qui fait que lorsque les cellules sont à confluence elles vont s’arrêter de proliférer
naturellement.
Toutes ces règles sont enfreintes par les cellules immortelles et transformées, les transformés
respectant vraiment rien du tout alors que les immortelles sont entre les deux mais ont un
nombre de division illimité.
Les cellules normales respectent toutes ces règles.
Intérêts de la culture cellulaire
C’est fondamentale pour regarder comment s’effectue la différenciation des cellules, études
sur la structures des cellules (fonctionnement du cytosquelette), grâce aux cellules on peut
faire des diagnostiques comme le caryotype qui est réalisé à partir de lymphocytes qui sont
mis en culture, on peut faire des recherches de mutations. On peut faire aussi de la thérapie :
greffe de moelle osseuse chez un patient qui n’a plus de moelle osseuse efficace donc pour lui
redonner des cellules du sang dont immunitaires. Si on pouvait amplifier les cellule en culture
on pourrait traiter plus de gens plus simplement mais on est souvent limité par la quantité de
matériel à greffer.
Exemple d’une culture fondamentale :
Ici ce sont des cultures de moelle osseuse humaine. Au bout d’un certain temps, si on fait un
marquage (anticorps) dirigé contre le facteur 8, on met en évidence toutes les cellules
exprimant le facteur 8 donc les cellules endothéliales donc on note dans cet amas de cellules
les facteurs 8 positives donc les cellules endothéliales. Si on prends un autre marqueur
endothéliale, on voit qu’il y a également du collagène 4 autour de ces cellules.
- On constate ici 4 cellules qui entourent une structure vide qu’on pourrait appelé une lumière
(vaisseaux) et on retrouve des éléments éparses qui sont de la matrice extracellulaire
(collagène 4).
- Enfin on fait une dernière série de marquage : On recherche l’actine alpha SN = spécifique
du muscle lisse, celle exprimé par les péricytes/ cellules musculaires lisses qui entourent les
cellules endothéliales dans les capillaires et on s’aperçoit qu’il y a des cellules musculaires
lisses au contact de cette structure endothéliale donc à partir de cette culture on observe des
cellules endothéliales qui sont alignés, un embryon de lumière, entouré d’une lame basale et
associé à des péricytes donc on a un vaisseau en culture qui se reconstitue.
- On récupère dans ce container tous les débris d’aspiration et faire une culture. L’intérêt c’est
de récupérer les cellules d’un patient pour savoir ce que sont ces cellules. On pourrait aider à
la définition anatomo-pathologique de la tumeur. On pourrait faire une recherche à partir
d’agents chimio thérapeutiques de ceux qui seraient efficace/inefficace sur ces cellules en
culture et on pourrait faire la corrélation chez le patient donc éviter de faire des traitements
sans effet. On est donc là au niveau d’une culture cellulaire qui est très proche de la thérapie.
Analyse des cellules.
- le FACS : cytomètre de flux.
C’est un appareil qui permet d’analyser une population cellulaire en suspension, pour cela les
cellules en suspension vont être séparés et mises chacune dans une micro goutte et passer
devant un laser. La lumière émise par le laser qui passe de l’autre côté est recueillis par des
détecteurs qui vont permettre d’évaluer la taille, la granulométrie de la cellule et si on a fait un
marquage des cellules fluorescent ça donne si la cellule est fluorescente ou non. C’est l’aspect
descriptif. Par ailleurs cet appareil est trieur c’est à dire qu’en fonction d’un paramètre :
 6
7
8
6
7
8
1
/
8
100%