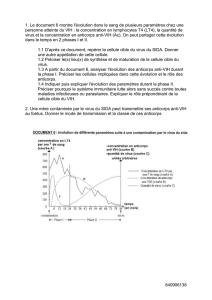
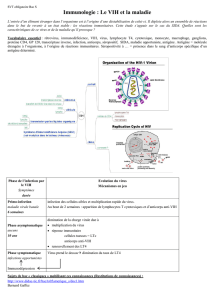
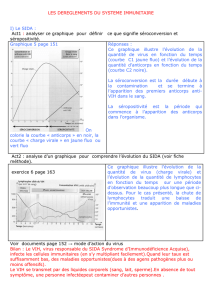
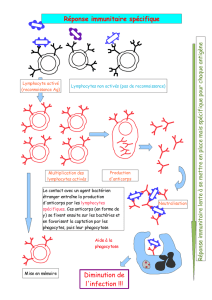
Mode d`administration

RA-2006/589a/cdm - 1 -
Résumé des Caractéristiques du Produit
1. DENOMINATION DU MEDICAMENT
HEPACAF 5000 I.U./100 ml, poudre et solvant pour solution pour perfusion.
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
HEPACAF 5000 I.U./100 ml est un concentré humain lyophilisé d’immuglobulines
spécifiques anti-hépatite B, préparé à partir d’un pool de plasma humain contenant
un titre élevé d’anticorps anti-hépatite B. HEPACAF 5000 I.U./100 ml est préparé à
partir de plasma humain provenant de donneurs bénévoles non rémunérés.
La teneur en protéines humaines s’élève à environ de 50 g/l dont au moins 95%
d’IgG. Les sous-classes d’immunoglobuline G (IgG) sont: IgG1: 57%, IgG2: 35%,
IgG3: 6%, IgG4: 2%.
La teneur d’immunoglobulines humaines spécifiques anti-hépatite B est de minimum
50 U.I./ml.
En ce qui concerne les adjuvants, voir 6.1.
3. FORME PHARMACEUTIQUE
Poudre et solvant pour solution pour perfusion.
4. DONNEES CLINIQUES
4.1 Indications thérapeutiques
Prévention d’une réinfection du greffon hépatique par l’hépatite B chez des patients
HBsAg-positifs (thérapie adjuvante en cas de transplantation du foie).
4.2 Posologie et mode d’administration
Posologie
Chez les adultes:
10.000 U.I. le jour de la transplantation, durant la chirurgie de greffe, suivies par
10.000 U.I. quotidiennement durant la première semaine qui suit la transplantation.

RA-2006/589a/cdm - 2 -
Par la suite, le dosage (quantité + moment) est adapté en fonction du titre d’anticorps
anti-HBs dans le sang (taux sérique protecteur au-delà de 100-150 U.I./l chez les
patients HBV-DNA-négatifs).
Chez les patients à risque (risque élevé de réinfection: statut de réplication virale
élevé, c’est-à-dire sérum testé positif pour l’Antigène HBe et/ou HBV-DNA,
détection HBc-Ag dans la biopsie du foie) le taux sérique protecteur recommandé est
supérieur à 500 mU.I./ml.
Ce schéma doit être suivi durant toute la vie du patient. L’expérience permet de
conclure que le nombre d’administrations diminue à partir de la deuxième année qui
suit l’année de transplantation.
Chez les enfants:
La posologie doit être adaptée à la surface corporelle à base de 10.000 U.I./1.73 m2.
Dans toutes ces circonstances, une vaccination contre le virus de l’hépatite B est
fortement recommandée. La première dose peut être administrée le même jour que
l’immunoglobuline humaine anti-hépatite B, toutefois dans des sites différents.
Mode d’administration
HEPACAF est présenté sous forme de poudre qui doit être dissoute avec de l’eau
pour injections, comme décrit dans 6.6 (“Instructions pour l’utilisation, la
manipulation et l’élimination”). Après reconstitution au moyen du solvant, la
préparation doit être administrée par voie intraveineuse.
Le débit sera adapté en fonction de la tolérance clinique, sans toutefois dépasser, au
cours de la première demi-heure, un débit 1 ml/kg/h. Si le produit est bien toléré le
débit peut être progressivement augmenté jusqu’à un maximum de 4 ml/kg/h.
4.3 Contre-indications
Hypersensibilité aux immunoglobulines humaines, en particulier chez les patients
qui souffrent d’une déficience en IgA, avec anticorps IgA.
Hypersensibilité à un ou plusieurs composants de la préparation, en particulier aux
traces de pepsine porcine qui sont utilisées dans le processus de fabrication.
4.4 Mises en garde spéciales et précautions particulières d’emploi
Les taux sériques d’anticorps anti-HBs doivent être contrôlés régulièrement chez les
patients.
La prudence est de rigueur chez les patients avec une hypersensibilité à un des
composants de la préparation.
Les patients doivent être observés au moins 20 minutes après l’administration.

RA-2006/589a/cdm - 3 -
Certains effets indésirables graves résultent d’une vitesse de perfusion trop élevée.
La vitesse de perfusion recommandée doit être strictement respectée. Durant la
période de perfusion, les patients doivent faire l’objet d’une étroite surveillance. En
cas de survenue d’effets indésirables, la vitesse de perfusion doit être adaptée et, si
nécessaire, la perfusion doit être arrêtée jusqu’à ce que ces effets aient disparus.
Il faut tenir compte de la teneur en sucrose (27,5 mg/ml) chez les patients présentant
un risque d’insuffisance rénale. Depuis la survenue de cas d’immunoglobulines
intraveineuses (IVIG) dans lesquels des facteurs de risque ont été identifiés, comme
une insuffisance rénale préexistante, du diabète mellitus, de l’hypovolémie, une
surcharge pondérale, la prise simultanée de produits médicinaux à toxicité rénale ou
à un âge de plus de 65 ans, il existe un risque théorique de lésion rénale chez les
patients qui sont traités avec des immunoglobulines anti-hépatite B (HepBIg). Tandis
que ces rapports sur les troubles rénaux et l’insuffisance rénale aiguë sont associés à
l’utilisation de nombreux des produits IVIG autorisés, les produits contenant du
sucrose comme stabilisateur, constituent une part disproportionnée du nombre total.
Pour tous les patients, l’administration de HepBIg nécessite:
- une hydratation suffisante avant l’administration de la perfusion avec HepBIg
- la surveillance du débit urinaire
- le contrôle des taux de créatine dans le sérum
- la prévention de l’administration simultanée de diurétiques de l’anse
En cas de lésion rénale, une interruption de l’administration de HepBIg doit être
envisagée.
Chez les patients à risque, l’utilisation de produits HepBIg qui ne contiennent pas de
sucrose, peut être envisagée.
Il faut tenir compte de la teneur en glucose (7.5 mg/ml) en cas de diabète latent, en
cas de diabète ou chez les patients qui suivent un régime pauvre en hydratés de
carbone. Ce produit doit être pris avec précaution chez les patients présentant un
diabète asymptomatique qui peuvent développer provisoirement une glycosurie et
chez les patients qui ne tolèrent pas l’un des constituants de la préparation.
Certains effets secondaires peuvent survenir plus souvent chez les patients
présentant une hypo- ou agammaglobulinémie avec ou sans déficience en IgA.
Des réactions d’hypersensibilité pure sont rares. Elles peuvent se produire dans de
rares cas de déficience IgA avec des anticorps anti-IgA ou d’hypersensibilité aux
traces de pepsine animale présente dans la préparation.
En cas de réaction anaphylactique ou choc, l’administration doit être interrompue
immédiatement. En cas de choc, il y a lieu d’instaurer un traitement classique de
choc.
Les mesures habituelles destinées à prévenir toute infection liée à l’administration
des médicaments préparés à partir de sang ou de plasma humain comprennent la
sélection des donneurs, le dépistage des marqueurs spécifiques d’agents infectieux
sur chaque don et sur le pool plasmatique ainsi que la mise en œuvre, dans le
procédé de fabrication d’étapes efficaces vis-à-vis de l’inactivation/élimination des

RA-2006/589a/cdm - 4 -
virus. Cependant le risque de transmission d’agents infectieux ne peut être
définitivement exclu lorsque sont administrés des médicaments préparés à partir de
sang ou de plasma humain. Ceci s’applique également aux virus inconnus ou
émergents et autres agents pathogènes.
Ces procédures sont considérées comme efficaces vis-à-vis de virus enveloppés tels
que le virus HIV, le virus HBV et le virus HCV. L’efficacité des ces procédures peut
être limitée vis-à-vis de virus non enveloppés tels que le virus HAV et le Parvovirus
B19.
L’expérience clinique est en faveur d’une absence de transmission de l’hépatite A ou
du parvovirus B19 avec les immunoglobulines. Il est également établi que la teneur
en anticorps apporte une contribution considérable à la sécurité virale.
Dans l’intérêt du patient il est fortement recommandé, lors de chaque administration
d’HEPACAF d’enregistrer le nom et numéro de lot du produit afin d’assurer un suivi
des lots utilisés.
4.5 Interaction avec d’autres médicaments et autres formes d’interactions
Interactions avec des virus vivants atténués
L’administration d’immunoglobulines peut diminuer l’efficacité d’une vaccination
par un virus vivant atténué tel que rougeole, oreillons, rubéole, varicelle, pour une
période de 6 semaines à 3 mois.
Il est recommandé d’attendre 3 mois, après l’administration d’immunoglobulines
humaines anti-hépatite B, avant de subir une vaccination par des virus vivants
atténués humaines.
Les immunoglobulines humaines anti-hépatite B doivent être administrées trois à
quatre semaines après la vaccination par un tel vaccin vivant atténué. Si
l’administration d’immunoglobulines humaines anti-hépatite B est nécessaire dans
les trois à quatre semaines après la vaccination, il faut effectuer un rappel trois mois
après l’administration des immunoglobulines humaines anti-hépatite B.
Interférence avec les analyses sérologiques
Après l’administration de cette préparation, les titres d’anticorps peuvent augmenter
temporairement en raison d’une transmission passive, ce qui peut donner lieu à des
résultats sérologiques faussement positifs.
La transmission passive d’anticorps aux antigènes d’érythrocytes, par exemple A, B,
D peut interférer avec certains tests sérologiques pour les anticorps des globules
rouges (par exemple, test Coombs).

RA-2006/589a/cdm - 5 -
4.6 Grossesse et allaitement
La sécurité d’HEPACAF 5000 I.U./100 ml pendant la grossesse n’a pas été établie par
des études cliniques contrôlées et il faut donc être prudent lors de l’administration à
des femmes enceintes et des mères qui allaitent.
Une expérience clinique de longue durée avec les immunoglobulines montre que
leur administration pendant la grossesse ne laisse présager aucun effet préjudiciable
pour le fœtus et le nouveau-né.
Les immunoglobulines sont des composants normaux issus du plasma humain: ils
passent dans le lait maternel et ne pourraient avoir aucun effet indésirable chez le
nouveau-né.
4.7 Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
Aucun effet sur l’aptitude à conduire des véhicules ou à utiliser des machines n’a été
observé.
4.8 Effets indésirables
Des effets secondaires suivants ont été enregistrés dans le cadre de la
pharmacovigilance:
Des maladies telles que frissonnements, fièvre, maux de tête, vomissements,
réactions allergiques, nausée, arthralgie et léger mal de dos peuvent parfois survenir.
Dans de rares cas, l’immunoglobuline humaine peut causer une hypotension et dans
quelques cas exceptionnels, un choc anaphylactique, même si le patient n’a présenté
aucune hypersensibilité lors d’administrations antérieures.
Pendant le traitement préventif d’une réinfection en cas de greffe, des cas très rares
de réaction d’intolérance peuvent être associés à une augmentation de l’intervalle
entre deux administrations.
Pour plus d’informations sur les agents transmissibles, voir le point 4.4.
4.9 Surdosage
Aucun cas de surdosage n’a été constaté à ce jour.
5. PROPRIETES PHARMACOLOGIQUES
5.1 Propriétés pharmacodynamiques
Catégorie pharmacothérapeutique: immunoglobulines spécifiques intraveineuses
anti-hépatite B. Code ATC: JO6BB04.
 6
7
8
6
7
8
1
/
8
100%