fièvre de west nile

414 Manuel terrestre de l’OIE 2008
CHAPITRE 2.1.20.
FIÈVRE DE WEST NILE
RÉSUMÉ
Le virus de la fièvre de West Nile (WNV pour West Nile virus) est un membre du genre Flavivirus
appartenant à la famille Flaviviridae. Cet arbovirus se maintient dans la nature en circulant de
manière cyclique chez les oiseaux et les moustiques ; de nombreuses espèces d’oiseaux et de
moustiques assurent la réplication de ce virus. Pour de nombreuses espèces aviaires, l'infection
par le WNV ne cause aucun signe manifeste tandis que d'autres oiseaux, tels que les corneilles
américaines (Corvus brachyrhynchos) et les geais bleus (Cyanocitta cristata), succombent souvent
à une maladie systémique mortelle. Parmi les mammifères, la maladie se déclare principalement
chez les chevaux et les humains.
Chez les chevaux, les signes cliniques induits par l'infection par le WNV correspondent à une
encéphalite ou à une encéphalomyélite virale. Les infections sont transmises par les moustiques et
sont saisonnières dans les climats tempérés, avec un pic au début de l’automne dans l'hémisphère
Nord. Les chevaux affectés montrent fréquemment une ataxie légère à sévère. Les signes cliniques
peuvent aller d’une incoordination légère au décubitus. Quelques chevaux présentent également
un affaiblissement, des tremblements musculaires, et des déficits des nerfs crâniens. La fièvre n'est
pas un facteur de la maladie uniformément présent chez les chevaux.
Identification de l'agent pathogène : les tissus aviaires contiennent généralement des
concentrations plus élevées de virus que les tissus équins. Le cerveau et la moelle épinière sont
les tissus de prédilection pour l'isolement du virus chez les chevaux. Chez les oiseaux, le rein, le
cœur, le cerveau, le foie ou l'intestin peuvent contenir le virus. Les cultures de cellules de rein de
lapin ou les cellules Vero sont le plus généralement employées pour l'isolement du virus. Le WNV
induit un effet cytopathogène (ECP) dans les cultures cellulaires sensibles à l’infection. L'acide
nucléique viral et les antigènes viraux peuvent être détectés dans les tissus des animaux infectés
par la technique de la transcription inverse couplée à une réaction d'amplification en chaîne par
polymérase (RT-PCR) et par immunohistochimie. La méthode la plus sensible pour identifier le
WNV dans les tissus équins est une RT-PCR nichée.
Épreuves sérologiques : les anticorps contenus dans le sérum équin peuvent être identifiés par
une méthode immuno-enzymatique (ELISA) de capture d'IgM ou d’IgG, une inhibition de
l’hémagglutination (IH), ou par une séroneutralisation par réduction de plages (PRN pour plaque
reduction neutralisation). Les méthodes ELISA et PRN sont les plus généralement employées pour
identifier les anticorps anti-WNV dans le sérum aviaire. Des réactions sérologiques croisées avec
des flavivirus apparentés, tels que le virus de l’encéphalite de St-Louis, le virus de l’encéphalite
japonaise ou l’encéphalite à tiques (TBE pour tick-borne encephalitis), peuvent survenir.
Spécifications applicables aux vaccins et aux produits biologiques à usage diagnostique :
un vaccin inactivé au formol et dérivé d’une culture de tissus, un vaccin vivant utilisant le
canarypoxvirus comme vecteur, un vaccin ADN et un vaccin chimère ont reçu une autorisation pour
utilisation chez les chevaux.
A. INTRODUCTION
Le virus de la fièvre de West Nile (WNV pour West Nile Virus) est un arbovirus transmis par les moustiques qui
appartient au genre Flavivirus de la famille Flaviviridae (26). Le genre Flavivirus contient également d’autres virus
tels que, notamment, ceux de l'encéphalite japonaise (voir le Chapitre 2.1.7.), de l’encéphalite de St-Louis, Le
virus de la vallée de Murray, le virus Usutu et le virus de Kunjin (6). Le WNV a une large répartition géographique

Chapitre 2.1.20. — Fièvre de West Nile
Manuel terrestre de l’OIE 2008 415
incluant des régions d'Europe, d’Afrique, d'Asie, d'Australie (virus Kunjin), d'Amérique du Nord, centrale et du
Sud. La dispersion ainsi que la réintroduction du virus à partir de secteurs enzootiques vers des régions qui
subissent des épizooties sporadiques s’effectueraient par les oiseaux migrateurs (6). Le WNV se maintient grâce
à un cycle de transmission moustique-oiseau-moustique, tandis que les humains et les chevaux sont des
impasses épidémiologiques. L'analyse génétique des isolats divise les souches en deux clades. Les isolats de la
lignée 1 sont isolés en Afrique centrale et du Nord, en Amérique du Nord et Centrale, en Colombie et en
Argentine en Amérique du Sud (18), en Australie (virus de Kunjin), en Europe, en Inde et en Israël. Les souches
de la lignée 2 sont enzootiques à Madagascar et en Afrique australe et centrale avec une cocirculation des deux
lignées du virus en Afrique centrale (3, 7). Récemment, un rapport a fait état de la présence de la lignée 2 en
Hongrie. Les souches de chaque lignée sont responsables des maladies humaines et animales tandis que les
épidémies humaines et équines récentes sont provoquées par des virus appartenant à la lignée 1.
Le virus de la fièvre de West Nile a été identifié comme étant un organisme pathogène humain en Afrique durant
la première moitié du 20e siècle. Bien que plusieurs épidémies de fièvre de West Nile aient été décrites,
l'encéphalite comme conséquence de l’infection humaine n’a que rarement été décrite avant 1996, mais depuis
lors, des épidémies d'encéphalite humaine causées par le WNV ont été rapportées en Roumanie, Russie, Israël,
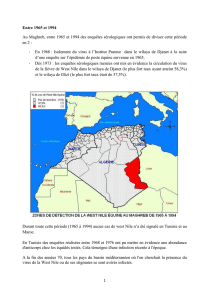
Amérique du Nord, France et Tunisie (4, 11, 13, 15, 33). Pendant les années 1960, la fièvre de West Nile chez
les chevaux a été signalée en Égypte et en France (23, 25). Depuis 1998, des manifestations de fièvre de West
Nile ont été rapportées en Italie, en France, au Canada, aux États-Unis d’Amérique, en Israël et au Maroc (8, 14,
19, 21). En occident, la répartition géographique du virus s’est nettement agrandie partant d'une petite région le
long de la côte Est de l'État de New York pour inclure les états contigus des États-Unis d'Amérique, le Canada, le
Mexique, les îles des Caraïbes, l’Amérique centrale, l’Argentine, la Colombie et le Venezuela (10, 18, 21, 30). En
dehors des États-Unis et du Canada, l’introduction du WNV dans les régions occidentales n’a pas été
caractérisée par de grands foyers de la maladie dans une espèce en particulier, ni par une mortalité significative ;
cela pourrait être dû à l’exposition auparavant à des flavivirus indigènes présents dans ces régions.
La période d'incubation de la fièvre de West Nile est estimée de 3 à 15 jours après la transmission par les
moustiques. Une virémie passagère accompagnée d’un faible titre de virus précède le début de la phase clinique
(5, 25). L’encéphalite due au WNV se produit seulement chez 1 % des chevaux infectés ; la majorité des chevaux
infectés ne présente pas de signes cliniques (21). La maladie chez les chevaux est fréquemment caractérisée par
une ataxie allant de légère à sévère. De plus, les chevaux peuvent présenter des signes de faiblesse, de
tremblements musculaires et des déficits des nerfs crâniens (8, 21, 22, 27). La fièvre n’est pas présente de
manière systématique. Le traitement est symptomatique et les signes cliniques peuvent progresser jusqu’à un
décubitus final. Le taux de mortalité est approximativement de 1 sur 3 chevaux cliniquement affectés. Le
diagnostic différentiel chez les chevaux inclut d'autres encéphalites arbovirales (par exemple, les
encéphalomyélites équines vénézuélienne, de l’Est ou de l’Ouest et l’encéphalite japonaise), la myélite équine
protozoaire (Sarcocystis neurona), l’herpesvirus équin 1, la maladie de Borna et la rage.
La plupart des espèces aviaires peuvent être infectées par le WNV ; les conséquences de la maladie clinique
sont variables. Les poulets et les dindes sont résistants. Des cas de maladie avec signes nerveux ont été
rapportés chez des oiseaux dans des zoos aux États-Unis et chez des oies domestiques en Israël et au canada
(1, 28, 33). Le WNV a été associé à une maladie sporadique dans un nombre restreint d'autres espèces,
notamment chez les écureuils, les tamias, les chauves-souris, les chiens, les chats, le cerf de Virginie, les rennes,
les moutons, les alpacas, les alligators et chez un phoque commun pendant des périodes d’intense activité virale
localement. La plupart des infections humaines se produisent lors d’une transmission normale par les
moustiques, mais des infections acquises en laboratoire ont été rapportées. Dans des cas cliniquement suspects,
les échantillons destinés au diagnostic de tous les animaux, en particulier les oiseaux, devraient être manipulés
dans un laboratoire de confinement de niveau 3 (voir le Chapitre 1.1.2., « Biosécurité et Biosûreté au laboratoire
de microbiologie vétérinaire et dans les animaleries ») (24). Chez l’homme, la transmission du WNV par
transfusion sanguine, greffe d’organes ou la tétée a été confirmée.
En raison de l'existence d’infections inapparentes par le WNV, les critères de diagnostic impliquent une
association de l’évaluation clinique et de l'analyse de laboratoire.
B. TECHNIQUES DE DIAGNOSTIC
1. Identification de l'agent pathogène
a) Culture
Les prélèvements pour l'isolement du virus comprennent le cerveau et la moelle épinière des chevaux
atteints d’encéphalites (21, 22) ; divers prélèvements de tissus d'oiseau comprenant le cerveau, le cœur ou
le foie peuvent également être utilisés avec succès (28). Le WNV a été isolé depuis le rein mais le tissue
peut être toxique pour les cultures cellulaires. En général, l’isolement du virus est obtenu plus facilement à

Chapitre 2.1.20. — Fièvre de West Nile
416 Manuel terrestre de l’OIE 2008
partir d’échantillons aviaires. Le virus peut être propagé sur des cultures de cellules sensibles, telles que les
cellules de rein de lapin (RK-13) et les cellules de rein du singe vert africain (Vero), ou encore des œufs
embryonnés de poulet. Les inoculations intracérébrales de souriceaux nouveau-nés sont moins efficaces
pour isoler le virus à partir de tissus de mammifères que les méthodes de culture de cellules. En culture de
cellules, plus d'un passage doit être effectué pour observer un effet cytopathogène (ECP). La confirmation
de l’isolement du WNV est réalisée par un marquage indirect par des anticorps fluorescents sur les cultures
de cellules infectées ou par des méthodes de détection de l’acide nucléique (voir ci-dessous).
b) Méthodes immunologiques
Le marquage immunohistochimique (IHC) de tissus aviaires après fixation au formol est une méthode fiable
pour l'identification de l'infection du WNV chez les oiseaux. Le cerveau, le cœur, les reins, la rate, le foie,
l’intestin et les poumons sont souvent positifs en IHC chez les oiseaux infectés. Le taux de réussite chez les
animaux positifs est augmenté par l’examen de plusieurs organes. La spécificité de l'identification (par
exemple, spécifique des flavivirus ou spécifique du WNV) dépend du choix de l'anticorps détecteur. Les
tissus du cerveau et de la moelle épinière des chevaux atteints d’une encéphalite due au WNV ne sont pas
positifs dans les tests IHC ; environ 50 % des cas de fièvre de West Nile équine donnent des résultats
faux-négatifs. L’absence d’identification d’un antigène du WNV dans le système nerveux central équin ne
permet pas d’exclure la présence de l'infection.
c) Méthodes d'identification de l’acide nucléique
La détection de l’acide nucléique par la technique de la transcription inverse couplée à une réaction
d'amplification en chaîne par polymérase (RT-PCR) augmente de manière significative l'identification des
tissus infectés par le WNV, plus particulièrement quand une PCR nichée est réalisée sur des échantillons
équins, frais et non fixés, de cerveau et de moelle épinière (16). La méthode de RT-PCR nichée pour
détecter l'acide nucléique du WNV codant une partie du gène E est décrite ci-dessous. Cette méthode,
développée en utilisant un isolat nord-américain de 1999, a permis de détecter l'ARN du WNV à partir de
tissus animaux lors de récentes épizooties nord-américaines. Le virus de l’encéphalite de St Louis n'est pas
détecté par cette méthode. Les virus de la fièvre de West Nile, appartenant à la lignée 1, de Chine, de
France, d'Égypte, d'Israël, d'Italie, du Kenya, du Mexique et de Russie montrent une séquence nucléotidique
fortement conservée dans la région ciblée, indépendamment de l'espèce d'origine (17). L'analyse de la
séquence pour la souche Ouganda 1937 (GenBank M12294), appartenant à la lignée 2, dans la région
visée par les amorces de PCR indique qu’il n’y aurait pas amplification pour les souches du WNV de la
lignée 2. D'autres virus du sérogroupe de l’encéphalite japonaise n'ont pas été examinés. Les méthodes non
nichées, y compris la PCR en temps réel, présentent moins de risques de contamination transversale en
laboratoire et peuvent être appliquées avec succès aux échantillons de tissus aviaires (17). Une PCR en
temps réel pour détecter l’acide nucléique du WNV a été décrite (29). Afin de normaliser les différentes
techniques moléculaires de diagnostic du WNV, une étude inter-laboratoire de compétence basée sur
l’examen de tissu fixé dans le formol a été mise en œuvre et elle a fait l’objet d’un rapport (20). Les tissus
choisis pour la PCR sont identiques à ceux choisis pour l’isolement viral.
• Technique de la transcription inverse couplée à une réaction d'amplification en chaîne niche par
polymérase (RT-nPCR)
La RT-nPCR décrite ici comprend plusieurs procédures : l’extraction de l’ARN, la transcription inverse pour
produire de l'ADN à partir de l’ARN et la première étape de la PCR, la deuxième étape de la PCR en
utilisant les amorces « nichées », et la détection de l'amplicon de taille appropriée par migration dans un gel
d'électrophorèse. Les régions de 445 et 248 pb (paire de bases) du gène codant la protéine E du WNV sont
respectivement amplifiées dans la première étape de la PCR et PCR nichée. Les kits de diagnostic et les
réactifs décrits ci-dessous sont fournis comme exemple. Des produits équivalents peuvent être disponibles
par d'autres sources. Une précaution extrême en manipulant tous les matériaux et l’utilisation de témoins
appropriés sont essentiels pour assurer des résultats précis. Les précautions à prendre ont été couvertes
dans le Chapitre 1.1.5., « Validation et contrôle qualité des méthodes d'amplification en chaîne par
polymérase (PCR) utilisées pour le diagnostic des maladies infectieuses ». Des échantillons en double de
chaque prélèvement devraient être traités et examinés pour augmenter la confiance des résultats de
l’analyse. Il faut respecter les précautions appropriées lors de manipulation de réactifs dangereux tels que le
bromure d'éthidium.
• Extraction de l’ARN viral
De 50 à 100 mg de tissus sont utilisés pour extraire l’ARN total avec le réactif Trizol® (Life Technologies,
Grand Island, NY, USA) selon les instructions du fabricant. Parallèlement de l’ARN total est extrait d’un
stock témoin du WNV contenant 10-100 DICT50 (dose de virus infectant 50 % de la culture tissulaire) pour
un volume de 100 µl.

Chapitre 2.1.20. — Fièvre de West Nile
Manuel terrestre de l’OIE 2008 417
• Transcription inverse et première étape PCR
Amorces de la première étape :
1401: 5’-ACC-AAC-TAC-TGT-GGA-GTC-3’
1845: 5’-TTC-CAT-CTT-CAC-TCT-ACA-CT-3’
i) Dissoudre les échantillons d’ARN extraits dans 12 µl d’eau purifiée de toute RNase.
ii) Incuber à 70 °C pendant 10 min.
iii) Ajouter 2 µl de chaque échantillon d’ARN dénaturé à 48 µl de mélange RT-PCR dont la composition
finale est de :
10 mM de Tris/HCl, pH 8,3
50 mM de KCl
2,0 mM de MgCl2
0,8 mM de mélange de déoxynucléoside triphosphate (dNTP)
25 unités de M-MLV (Moloney murine leukaemia virus) RT
1,25 unités d’inhibiteur de RNase
1,25 unités d’AmpliTaq GoldTM (Applied Biosystems, Foster City, CA, USA)
37,5 pmol d’amorces de la première étape.
Inclure des témoins négatifs en utilisant 2 µl d’eau purifiée de toute RNase à la place de l’ARN
dénaturé.
iv) Incuber les tubes de réaction à 45 °C pendant 45 min.
v) Incuber les tubes de réaction à 95 °C pendant 11 min.
vi) PCR de 35 cycles :
Dénaturation à 95 °C pendant 30 s
Hybridation des amorces à 55 °C pendant 45 s
Elongation des amorces à 72 °C pendant 60 s, (pour le 35e cycle, élongation des amorces à 72 °C
pendant 5 min).
vii) Laisser les échantillons à 4 °C jusqu’à la deuxième étape de la PCR.
• Seconde étape PCR (nichée)
Amorces de la deuxième étape:
1485 : 5’-GCC-TTC-ATA-CAC-ACT-AAA-G-3’
1732 : 5’-CCA-ATG-CTA-TCA-CAG-ACT-3’
i) Pour chaque échantillon et témoin, ajouter 1,5 µl du produit de la première étape d’amplification dans :
48,5 µl du mélange de PCR avec une composition finale de :
10 mM de Tris/HCl, pH 8,3
50 mM de KCl
2,0 mM de MgCl2
0,8 mM d’un mélange de déoxynucléoside triphosphate (dNTP)
1,25 unités d’AmpliTaq GoldTM (Applied Biosystems, Foster City, CA, USA)
37,5 pmol de chaque amorce niche.
ii) Incuber les tubes de réaction à 95 °C pendant 11 min.
iii) PCR de 35 cycles :
Dénaturation à 95 °C pendant 30 s
Hybridation des amorces à 55 °C pendant 45 s
Elongation des amorces à 72 °C pendant 60 s (pour le 35e cycle, élongation de l’amorce à 72 °C
pendant 5 min).
iv) Laisser les échantillons à 4 °C ou –20 °C jusqu’à l’électrophorèse.
• Analyse des produits de PCR par migration en gel d’électrophorèse
i) Préparer une solution d’agarose 2,5 % NuSieve® 3/1 (FMC Bioproducts, Rockland, Maine, USA) dans
du Tris/borate 0,045 mM, pH 8,6, EDTA 1,5 mM (acide tétra-acétique de diamine d'éthylène)
(× 1 tampon TBE). Faire bouillir l'agarose sur une plaque chauffante ou dans un four à micro-ondes
jusqu'à ce qu’elle soit complètement dissoute. Refroidir l'agarose à 45-55 °C. Ajouter 5 µl de bromure
d'éthidium (10 mg/ml) pour 100 ml d'agarose chaude et couler le gel d'agarose avec un peigne. Laisser
solidifier et ensuite retirer le peigne.

Chapitre 2.1.20. — Fièvre de West Nile
418 Manuel terrestre de l’OIE 2008
ii) Ajouter 30 µl de la solution de bromure d'éthidium (10 mg/ml) pour 600 ml du tampon TBE × 1. Placer
le gel dans l’appareil d’électrophorèse et remplir le réservoir de tampon.
iii) Mélanger 15 µl de chaque échantillon et témoin avec 5 µl de solution de chargement du gel (par
exemple, produit G-2526, Sigma, St Louis, MO, USA). Ajouter le marqueur de poids moléculaire de
100 pb (par exemple, Life technologies, Grand Island, NY, produit 15268-019, USA, s'étendant de 100
à 1 500 pb) dans au moins un puits du gel. Charger les échantillons dans les puits d'agar et lancer
l’électrophorèse à 65-75 volts jusqu'à ce que le colorant de chargement de gel ait parcouru
approximativement 2/3 de la longueur du gel.
iv) Visualiser et photographier le gel sous une lumière ultraviolette.
• Interprétation du résultat
Pour que l’analyse par PCR soit valide, les témoins positifs doivent montrer une bande de taille appropriée
(248 pb). Les témoins « no ARN » ne devraient pas présenter de bande. Les échantillons sont considérés
comme positifs s'il y a une bande de la même taille que le témoin positif. Les échantillons doubles doivent
présenter la même réaction. S'il y a une disparité, l'analyse devrait être répétée, en commençant par
l'extraction à partir du tissu. Si davantage de validation est exigée, le produit final de la PCR nichée peut
être séquencé et comparé aux séquences du WNV publiées et disponibles dans GenBank.
2. Épreuves sérologiques
Les anticorps peuvent être identifiés dans le sérum équin par technique immuno-enzymatique (ELISA) de capture
IgM, par inhibition de l’hémagglutination (IH), par ELISA IgG ou par séroneutralisation par réduction des plages
(PRN) (2, 12). L’ELISA de capture IgM décrit ci-dessous est particulièrement utile pour détecter les anticorps
résultant d’une exposition naturelle et récente au WNV. Les IgM équines spécifiques du WNV sont habituellement
détectables à partir de 7 à 10 jours jusqu’à 1 à 2 mois après l’infection. La plupart des chevaux sont positifs en
ELISA de capture IgM pour la fièvre de West Nile lorsque les premiers signes cliniques sont observés. Les
anticorps neutralisant le WNV sont détectables dans les sérums équins, 2 semaines après l’infection et peuvent
persister pendant plus d’une année. Les méthodes d’IH et de PRN sont le plus généralement employées pour
identifier les anticorps anti-WNV dans les sérums aviaires. Dans certaines analyses sérologiques, des réactions
croisées avec des flavivirus apparentés, tels que le virus de l'encéphalite de St Louis ou le virus de l'encéphalite
japonaise, surviendront. L'analyse par PRN est la plus spécifique parmi les analyses sérologiques du WNV ;
quand nécessaire, les titres en anticorps dirigés contre des flavivirus apparentés peuvent être examinés en
parallèle dans le sérum. En conclusion, l'historique de la vaccination contre le WNV doit être considérée dans
l'interprétation des résultats de la sérologie, en particulier dans l'analyse PRN et l’ELISA IgG. L’ELISA de capture
IgM peut être employé pour examiner les espèces aviaires ou autres à condition que l'anticorps de capture
spécifique de l'espèce soit disponible (par exemple, anti-poulet IgM). L'analyse par PRN est applicable sur
n'importe quelle espèce, y compris les oiseaux.
a) ELISA de capture IgM équine
Le WNV et des antigènes témoins négatifs pour l’ELISA de capture IgM peuvent être préparés à partir du
cerveau de souris (voir le Chapitre 2.5.5.), de culture de tissus ou de lignées de cellules recombinées (9).
Les sources commerciales des réactifs d'analyse du WNV sont disponibles en Amérique du Nord. Le sérum
témoin équin, bien qu’il ne soit pas un étalon international, peut être obtenu à partir des laboratoires
nationaux des services vétérinaires, Ames, Iowa, États-Unis. Le virus et les antigènes témoins négatifs
devraient être préparés en parallèle pour une analyse par ELISA. Les préparations d'antigène doivent être
titrées avec des sérums témoins pour optimiser la sensibilité et la spécificité de l'analyse. Les échantillons
de sérum équin sont examinés à une dilution de 1/400 et les échantillons équins de liquides céphalo-
rachidiens sont examinés à une dilution de 1/2. Pour assurer la spécificité, chaque échantillon de sérum est
examiné pour sa réactivité avec l'antigène viral et l'antigène témoin.
• Protocole
i) Ajouter sur des plaques ELISA de 96 puits à fond plat (par exemple, Immulon 2HB, Dynex
Technologies, Chantilly, VA, USA) 100 µl/puits d’IgM anti-équin diluées dans une solution tampon de
carbonate 0,5 M, pH 9,6, selon la dilution suggérée par le fabricant pour l'utilisation comme anticorps
de capture.
ii) Incuber les plaques durant une nuit à 4 °C en chambre humide. Les plaques sensibilisées peuvent être
conservées pendant plusieurs semaines.
iii) Avant l'utilisation, les plaques doivent être lavées 2 fois avec 200 à 300 µl/puits de solution
physiologique tamponnée au phosphate (PBS) 0,01 M, pH 7,2, contenant 0,05 % de Tween 20
(PBST).
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%