Chimie Organique : Structure, Stéréochimie, Alcanes, Alcènes
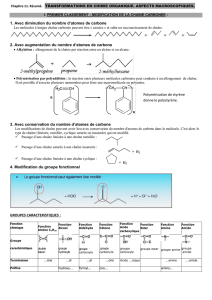
Telechargé par
Ikram Belyamani

1
Sommaire
Partie I : Structure et stéréochimie
I- Généralités .............................................................................................................................................................................................. 4
II- Représentations des molécules .............................................................................................................................................................. 6
1- Formules Planes : Formule semi-développée et développée .................................................................................................... 6
2- Isomérie plane .......................................................................................................................................................................... 6
a- Définition ...................................................................................................................................................................................... 6
b- Isomérie de chaine ........................................................................................................................................................................ 6
c- Isomérie de fonction ..................................................................................................................................................................... 6
d- Isomérie de position ..................................................................................................................................................................... 7
3- Degré d’insaturation ................................................................................................................................................................ 7
III- Stéréochimie ........................................................................................................................................................................................... 8
1- Introduction.............................................................................................................................................................................. 8
2- Représentations conventionnelles des molécules organiques ................................................................................................... 8
a- Convention de Cram ..................................................................................................................................................................... 8
b- Représentation perspective (ou perspective cavalière) .............................................................................................................. 8
c- Représentation Newman .............................................................................................................................................................. 9
d- Projection de Fischer .................................................................................................................................................................... 9
3- Stéréoisomérie ....................................................................................................................................................................... 10
a- Isomérie de conformation .......................................................................................................................................................... 10
b- Isomérie optique et énantiomérie .............................................................................................................................................. 11
Chiralité ........................................................................................................................................................................................ 11
Activité optique .......................................................................................................................................................................... 12
Enantiomérie .............................................................................................................................................................................. 13
Règles de Cahn-Ingold-Prelog (C.I.P.) ..................................................................................................................................... 14
Détermination de la configuration absolue ............................................................................................................................... 15
c- Diastéréoisomérie ....................................................................................................................................................................... 15
Molécules contenant plusieurs carbones asymétriques ............................................................................................................ 16
Nomenclature érythro et thréo ................................................................................................................................................... 17
Cas de molécules symétriquement substituées ......................................................................................................................... 17
IV- Effet électronique ................................................................................................................................................................................. 19
1- Effet inductif .......................................................................................................................................................................... 19
a- Les effets inductifs donneurs +I .............................................................................................................................................. 20
b- les effets inductifs attracteurs -I .............................................................................................................................................. 20
2- Effet mésomère ...................................................................................................................................................................... 20
a- Effet mésomère donneur + M .................................................................................................................................................... 20
b- Effet mésomère attracteur - M ................................................................................................................................................... 21
c- Les principaux systèmes conjugués .......................................................................................................................................... 21
V- Les intermédiaires réactionnels ........................................................................................................................................................... 22
1- Les carbocations .................................................................................................................................................................... 22
2- Les carbanions ....................................................................................................................................................................... 22
3- Les radicaux libres ................................................................................................................................................................. 23
Les Alcanes ................................................................................................................................................................................................. 26
I- Généralités ............................................................................................................................................................................................ 26
1- Définition ............................................................................................................................................................................... 26
2- Propriétés physiques .............................................................................................................................................................. 26
II- Représentations ..................................................................................................................................................................................... 27
III- Nomenclature ........................................................................................................................................................................................ 27
1- Alcanes à chaîne carbonée linéaire (sans ramification) .......................................................................................................... 27
2- Alcanes à chaîne carbonée ramifiée ....................................................................................................................................... 28
IV- Réactivité .............................................................................................................................................................................................. 28
1- Réaction avec les halogènes ................................................................................................................................................... 28
Les Alcènes ................................................................................................................................................................................................. 30
I- Généralités ............................................................................................................................................................................................ 30
II- Représentation ...................................................................................................................................................................................... 30
III- Nomenclature ........................................................................................................................................................................................ 30

2
IV- Propriétés Chimiques ........................................................................................................................................................................... 30
Les alcynes .................................................................................................................................................................................................. 35
I- Généralités ............................................................................................................................................................................................ 35
II- Nomenclature ........................................................................................................................................................................................ 35
III- Réactivité .............................................................................................................................................................................................. 35
1- Hydratation des alcynes ......................................................................................................................................................... 36
2- Réaction de réductions ........................................................................................................................................................... 36
Halogénures d’alkyles ................................................................................................................................................................................ 36
I- Généralités ............................................................................................................................................................................................ 37
II- Nomenclature ........................................................................................................................................................................................ 37
III- Réactivité .............................................................................................................................................................................................. 37
1- Substitutions nucléophiles ..................................................................................................................................................... 37
2- Eliminations ........................................................................................................................................................................... 38
Les Aromatiques ......................................................................................................................................................................................... 39
I- Généralités ............................................................................................................................................................................................ 39
II- Réactivité .............................................................................................................................................................................................. 39
1- Alkylation .............................................................................................................................................................................. 39
2- Acylation ............................................................................................................................................................................... 40
3- Nitration ................................................................................................................................................................................. 40
4- Polysubstitutions .................................................................................................................................................................... 40
5- Oxydation des chaînes latérales ............................................................................................................................................. 42
Les Alcools .................................................................................................................................................................................................. 44
I- Généralités ............................................................................................................................................................................................ 44
II- Nomenclature ........................................................................................................................................................................................ 45
III- Réactivité .............................................................................................................................................................................................. 45
1- Déshydratation des alcools..................................................................................................................................................... 45
2- Oxydation des alcools ............................................................................................................................................................ 45
Les Amines .................................................................................................................................................................................................. 47
I- Généralités ............................................................................................................................................................................................ 47
II- Nomenclature ........................................................................................................................................................................................ 47
III- Réactivité .............................................................................................................................................................................................. 48
1- Alkylation .............................................................................................................................................................................. 48
2- Elimination d'Hofmann .......................................................................................................................................................... 48
3- Acylation ............................................................................................................................................................................... 49
4- Réactions sur les carbonylés .................................................................................................................................................. 49
Les composés carbonylés ........................................................................................................................................................................... 50
I- Généralités ............................................................................................................................................................................................ 50
II- Nomenclature ........................................................................................................................................................................................ 51
III- Réactivité .............................................................................................................................................................................................. 52
1- Réductions ............................................................................................................................................................................. 52
Acides Carboxyliques ................................................................................................................................................................................. 53
I- Généralités ............................................................................................................................................................................................ 53
II- Nomenclature ........................................................................................................................................................................................ 53
III- Réactivité .............................................................................................................................................................................................. 53
1- Estérification .......................................................................................................................................................................... 53
2- Décarboxylation ..................................................................................................................................................................... 54
Travaux dirigés ........................................................................................................................................................................................... 56

3

4
I- Généralités
1- Débuts de la chimie organique
Au début du 19e siècle, Berzelius distinguait deux domaines de la chimie : la chimie minérale
(inorganique) et la chimie organique. La chimie organique s’occupait des composés synthétisés par
les organismes vivants comme les glucides, les lipides, les protéines, l’éthanol.... Contrairement aux
composés organiques, on savait déjà synthétiser un grand nombre de composés inorganiques. À
cette époque, on pensait que la synthèse des composés organiques nécessitait la présence d’une
« force vitale » qui existe seulement à l’intérieur des organismes.
En 1828, la synthèse de l’urée par Friedrich Wöhler à partir de composés inorganiques entama
une révolution en chimie organique. En effet, cette première synthèse d’un composé organique au
laboratoire montrait que la « force vitale » n’existe pas. Néanmoins, on a gardé jusqu’à nos jours la
division de la chimie en chimie organique et en chimie minérale. Toutefois, aujourd’hui la chimie
organique est définie de façon différente.
La chimie organique est la chimie de l’élément carbone
En effet, les composés organiques sont des composés principalement constitués de carbone lié à
d’autres éléments comme l’hydrogène, l’oxygène, l’azote, le chlore, le brome, le phosphore, le
soufre... Dans les molécules organiques, l’hydrogène est un élément très abondant, tandis que tous
les autres éléments n’apparaissent souvent qu’en traces.
Le nombre de composés de la chimie minérale (environ 100000) paraît minime par rapport aux
10 millions de composés organiques connus de nos jours.
2- Domaines d’application
La chimie organique joue le rôle principal dans l’industrie chimique. Ainsi les industries
pharmaceutique, cosmétique, alimentaire, l’industrie des plastiques...utilisent surtout des produits
organiques.

5
La chimie organique est partout
Molécules organiques sont l’essence de la vie (protéines, glucides, acides nucléiques...)
ADN : Composé de 4 bases avec une double hélice constituée de 4 acides aminés :
3- Les objectifs fondamentaux
L’objectif premier est de déterminer la relation entre la structure des molécules et ses propriétés
chimiques, physiques ou biologiques.
Le second est de comprendre le mécanisme des réactions pour cela on étudiera les différentes étapes
des réactions. Il sera ainsi possible de prévoir des réactions, le but étant en particulier d’augmenter
le nombre de synthèses organiques.
4- Eléments des composés organiques
Les éléments constitutifs des molécules organiques sont, par ordre de fréquence décroissant : Les
quatre éléments : C, H, O, N, les halogènes tels que : F, Cl, Br, I, le soufre.
Les différents types de carbone de la chimie organique
Un atome de carbone est dit :
primaire s'il est lié à un seul autre atome de carbone,
secondaire s'il est lié à deux autres atomes de carbone,
tertiaire s'il est lié à trois autres atomes de carbone,
quaternaire s'il est lié à quatre autres atomes de carbone.
Exemple :
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
1
/
70
100%