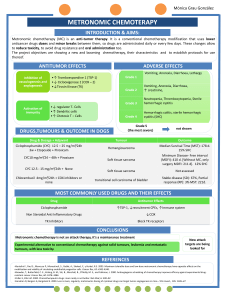
Euro Ewing 2012: Ewing's Sarcoma Clinical Trial Protocol
Telechargé par
elbessimeriem

International Randomised Controlled Trial for the
Treatment of Newly Diagnosed Ewing's Sarcoma Family
of Tumours
Euro Ewing 2012
Version 5.0
2nd June 2017
Coordinating Sponsor: University of Birmingham
Sponsor protocol number: RG_11-152
CAS code: SA3008
EudraCT number: 2012-002107-17
ISRCTN reference number: ISRCTN 92192408
This project has received funding from the European Union’s Seventh Framework Programme for
research, technological development and demonstration under grant agreement no °602856.

Euro Ewing 2012 Protocol
Page 2 of 80
Euro Ewing 2012 protocol_version 5.0_2-Jun-2017
CRCTU-PRT-QCD-001, version 1.0
TABLE OF CONTENTS
Table of contents ................................................................................................................................... 2
Introductory Pages ................................................................................................................................ 6
SIGNATURE PAGE ................................................................................................................................ 9
Amendments ........................................................................................................................................ 10
Trial Synopsis ...................................................................................................................................... 12
Trial Schema ........................................................................................................................................ 15
Abbreviations ....................................................................................................................................... 16
1. Background and Rationale ............................................................................................................ 18
1.1 Background............................................................................................................................ 18
1.1.1 Characterisation of ESFT .................................................................................................. 18
1.1.2 Treatment results in localised disease .............................................................................. 18
1.1.3 Treatment results in patients with primary pulmonary metastases and regional lymph
nodes ........................................................................................................................................... 18
1.1.4 Treatment results in patients with disseminated disease ................................................. 19
1.1.5 The value of bisphosphonates in the treatment of ESFT .................................................. 19
1.2 Trial Rationale........................................................................................................................ 20
1.2.1 Rationale for an international study ................................................................................... 20
1.2.2 Rationale for a VIDE and VAC/VAI versus VDC/IE/VC randomisation ............................. 21
1.2.3 Rationale for zoledronic acid randomisation ..................................................................... 22
2. Objectives and Outcome Measures ............................................................................................. 22
2.1 Objectives .............................................................................................................................. 22
2.2 Outcome Measures ............................................................................................................... 23
2.2.1 Primary outcome measure ................................................................................................ 23
2.2.2 Secondary outcome measures .......................................................................................... 23
3. Trial Design ..................................................................................................................................... 23
4. Eligibility .......................................................................................................................................... 23
4.1 Randomisation R1 ................................................................................................................. 23
4.2 Randomisation R2 ................................................................................................................. 24
5. Screening and Consent ................................................................................................................. 24
5.1 Screening ............................................................................................................................... 24
5.2 Informed Consent .................................................................................................................. 24
6. Randomisation................................................................................................................................ 25
6.1 Randomisation R1 ................................................................................................................. 25
6.2 Randomisation R2 ................................................................................................................. 27
6.3 Procedure for online randomisation ...................................................................................... 29
6.4 Emergency randomisation ..................................................................................................... 30
7. Treatment details ............................................................................................................................ 30
7.1 Investigational Medicinal Products (IMPs) ............................................................................ 30
7.2 Arm A treatment schedule ..................................................................................................... 31
7.2.1 Arm A: Overview ................................................................................................................ 31
7.2.2 Arm A: VIDE chemotherapy .............................................................................................. 32
7.2.3 Arm A: VAI chemotherapy ................................................................................................. 33
7.2.4 Arm A: VAC chemotherapy ............................................................................................... 34
7.2.6 Arm A: BuMel ..................................................................................................................... 35
7.2.7 Arm A: Zoledronic acid treatment ...................................................................................... 37
7.3 Arm B treatment schedule ..................................................................................................... 38

Euro Ewing 2012 Protocol
Page 3 of 80
Euro Ewing 2012 protocol_version 5.0_2-Jun-2017
CRCTU-PRT-QCD-001, version 1.0
7.3.1 Arm B: Overview ................................................................................................................ 38
7.3.2 Arm B: VDC/IE chemotherapy ........................................................................................... 39
7.3.3 Arm B: IE/VC chemotherapy ............................................................................................. 40
7.3.4 Arm A: BuMel ..................................................................................................................... 41
7.3.5 Arm B: Zoledronic acid treatment ...................................................................................... 41
7.4 Dose Modifications ................................................................................................................ 41
7.4.1 VIDE/VAI/VAC chemotherapy (Arm A) .............................................................................. 41
7.4.2 VDC/IE/VC chemotherapy (Arm B) ................................................................................... 44
7.4.3 Zoledronic acid (Arms A and B) ......................................................................................... 46
7.5 Treatment Compliance .......................................................................................................... 46
7.6 IMP Handling ......................................................................................................................... 46
7.7 Supportive Treatment ............................................................................................................ 46
7.7.1 Venous Access .................................................................................................................. 46
7.7.2 Antiemetics ........................................................................................................................ 46
7.7.3 Neutropenia ....................................................................................................................... 47
7.7.4 Blood products ................................................................................................................... 47
7.7.5 Pneumocystis carinii infection prophylaxis ........................................................................ 47
7.7.6 Hydration ........................................................................................................................... 47
7.8 Concomitant Medication ........................................................................................................ 47
7.9 Patient Follow Up .................................................................................................................. 47
7.10 Patient Withdrawal ................................................................................................................. 48
7.10.1 Withdrawal from Euro Ewing 2012 trial treatment ............................................................. 48
7.10.2 Withdrawal of consent to data collection ........................................................................... 48
7.10.3 Loss to follow-up ................................................................................................................ 48
8. Patient Monitoring and Assessment ............................................................................................ 48
8.1 Overview – Schedule of assessments .................................................................................. 49
8.2 Assessments at diagnosis ..................................................................................................... 50
8.2.1 Basic patient information ................................................................................................... 50
8.2.2 Blood chemistry ................................................................................................................. 50
8.2.3 Haematology ...................................................................................................................... 50
8.2.4 Radiological assessments ................................................................................................. 50
8.2.5 Other assessments ............................................................................................................ 50
8.2.6 Estimation of tumour involvement ..................................................................................... 50
Primary tumour volume ......................................................................................................................... 50
8.2.7 Diagnosis of ESFT ............................................................................................................. 51
8.2.8 Definition of pulmonary/pleural metastatic disease ........................................................... 51
8.2.9 Regional lymph node involvement..................................................................................... 52
8.3 Assessments during treatment .............................................................................................. 52
8.3.1 Prior to each cycle of chemotherapy ................................................................................. 52
8.3.2 Cardiac assessments ........................................................................................................ 52
8.3.3 Radiological assessments ................................................................................................. 52
8.3.4 Bone Marrow assessments ............................................................................................... 53
8.4 Assessments at the end of treatment .................................................................................... 53
9. Biological Studies .......................................................................................................................... 54
10. Adverse Event Reporting .............................................................................................................. 56
10.1 Reporting Requirements ........................................................................................................ 56
10.1.1 Adverse Events and Adverse Reactions ........................................................................... 56
10.1.2 Serious Adverse Events .................................................................................................... 56

Euro Ewing 2012 Protocol
Page 4 of 80
Euro Ewing 2012 protocol_version 5.0_2-Jun-2017
CRCTU-PRT-QCD-001, version 1.0
10.1.3 Reporting period ................................................................................................................ 57
10.1.4 Post study SARs and SUSARs: ........................................................................................ 57
10.2 Reporting Procedure ............................................................................................................. 57
10.2.1 Site ..................................................................................................................................... 57
10.2.2 UK Coordinating Centre .................................................................................................... 58
10.2.3 Reporting to the Competent Authority and Research Ethics Committee .......................... 58
10.2.4 Investigators ...................................................................................................................... 59
10.2.5 Data Monitoring Committee ............................................................................................... 59
11. Data Handling and Record Keeping ............................................................................................. 60
11.1 Data Collection ...................................................................................................................... 60
11.2 Archiving ................................................................................................................................ 60
12. Quality Management ...................................................................................................................... 60
12.1 Site Set-up and Initiation ....................................................................................................... 60
12.2 On-site Monitoring ................................................................................................................. 61
12.3 Central Monitoring ................................................................................................................. 61
12.4 Audit and Inspection .............................................................................................................. 61
12.5 Notification of Serious Breaches ........................................................................................... 61
13. End of Trial Definition .................................................................................................................... 62
14. Statistical Considerations ............................................................................................................. 62
14.1 Definition of Outcome Measures ........................................................................................... 62
14.1.1 Primary outcome measure ................................................................................................ 62
14.1.2 Secondary outcome measures .......................................................................................... 62
14.2 Induction/consolidation chemotherapy randomisation: general principles ............................ 63
14.3 Sample Size Calculations ...................................................................................................... 64
14.3.1 Induction/consolidation randomisation (R1) ...................................................................... 64
14.3.2 Zoledronic acid randomisation (R2)................................................................................... 65
14.4 Analysis of Outcome Measures ............................................................................................. 66
14.5 Planned Subgroup Analyses ................................................................................................. 68
14.6 Planned Interim Analysis ....................................................................................................... 68
14.7 Planned Main Analyses ......................................................................................................... 68
15. Trial Organisational Structure ....................................................................................................... 68
15.1 Coordinating-sponsor ............................................................................................................ 69
15.2 National Coordinating Centres .............................................................................................. 69
15.3 Trial Management Group ....................................................................................................... 69
15.4 Trial Steering Committee ....................................................................................................... 69
15.5 Data Monitoring Committee ................................................................................................... 69
15.6 Finance .................................................................................................................................. 70
16. Ethical Considerations .................................................................................................................. 70
17. Confidentiality and Data Protection ............................................................................................. 71
18. Insurance and Indemnity ............................................................................................................... 71
19. Publication Policy .......................................................................................................................... 71
20. Reference List ................................................................................................................................. 73
Appendix 1 – United Kingdom Specific Quality and Trial management Plan ............................... 76
Records of Screening/enrolment ....................................................................................................... 76
Informed Consent Form Review ........................................................................................................ 76
Site Set-up and Initiation .................................................................................................................... 76
Pharmacy ........................................................................................................................................... 76
On-site Monitoring .............................................................................................................................. 76

Euro Ewing 2012 Protocol
Page 5 of 80
Euro Ewing 2012 protocol_version 5.0_2-Jun-2017
CRCTU-PRT-QCD-001, version 1.0
Serious Breach Notification ................................................................................................................ 77
Archiving ............................................................................................................................................ 77
Appendix 2 – Definition of Adverse Events ...................................................................................... 78
Appendix 3 – Common Terminology Criteria for Adverse Events ................................................. 80
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
1
/
80
100%