Chimie : Réactions Acido-Basiques - Exercices et Corrigés MPSI
Telechargé par
sonia.garrouch

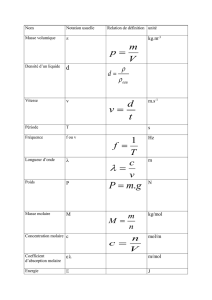
CHIMIE
T.D.
MP* Lycée Masséna - Académie de Nice
Chimie MPSI
Réactions acido-basiques
diag. de prédominances, dosages, pH
Remarque : dans tout le TD, en particulier les corrigés, on
utilisera la notation usuelle h= [H3O+] = [H+].
Exercice 1 - Solution d’acide phosphorique
L’acide phosphorique H3PO4est un triacide dont les 3
pKAvalent respectivement 2,2 , 7,2 et 12,3.
Q.1 Tracer le diagramme de prédominance en faisant ap-
paraître les domaines de majorité des différentes espèces.
Q.2 Une solution de NaH2PO4à5.10−2mol.l−1a un pH
de 4,7. Calculer la concentration des différentes espèces et
classer celles-ci selon leur importance relative.
Corrigé
R.1 Le domaine de prédominance de la base (resp. de
l’acide) correspond à pH > pkA+1(resp. pH < pkA−1).
On obtient donc, en notant A =PO4:
pH
2,2 7,2 12,3
H3AH2A−HA2−A3−
R.2 On est dans le domaine de prédominance de ) H2A−;
on a donc :
[H2A−]≃5.10−2mol.l−1(majoritaire)
[H3A] = [H2A−]h
KA1 ≃1,6.10−4mol.l−1(minoritaire)
[HA2−] = KA2[H2A−]
h≃1,6.10−4mol.l−1(minoritaire)
[A3−] = KA3[HA2−]
h≃4,0.10−12 mol.l−1(ultra-min.)
Exercice 2 - pH d’une solution d’acide chlorhy-
drique
On dispose d’une solution d’HCl à c0=10−4mol.l−1.
Q.1 Déterminer son pH.
On ajoute 0,1 ml de cette solution à 1 l d’eau distillée.
Q.2 Déterminer à nouveau le pH.
Corrigé
R.1 L’acide est totalement dissocié (acide fort) ; en négli-
geant l’apport d’ions H+par la réaction d’auto-protolyse
de l’eau on a ainsi pH =pc0=4. On doit vérifier
cette hypothèse : il faut que h≫[OH−]; soit en écrivant
h > 10[OH−]↔h2> 10Ke↔pH < 6,5 ce qui est bien le
cas.
R.2 La dilution donne une concentration en acide de c1=
10−8mol.l−1. L’application de la relation précédente pH =
pc1=8est cette fois-ci incorrecte. On ne peut évidemment
plus négliger l’apport des ions H+par l’eau ; on a alors :
h=c0+Ke
h⇔h=
c1+qc2
1+4Ke
2⇔pH =6,98
Pour bien visualiser la validité de l’approximation, on a
tracé ci-dessous le pH en fonction de la concentration cde
l’acide fort.
A retenir : pour pH < 6,5,pH =pc0où c0est la concen-
tration de l’acide fort.
Exercice 3 - pH d’une solution d’acide fluorhy-
drique
On dispose d’une solution de HF de pKA=3, 2 et dont la
concentration est notée c.
Q.1 Calculer le pH pour c=10−1mol.l−1.
Q.2 Calculer le pH pour c=10−5mol.l−1. Quelle loi est-
elle illustrée par ce cas ?
Corrigé
R.1 On écrit la RPE (non quantatitative) qui correspond
à l’action de l’acide faible sur l’eau ; celle-ci étant supposée
peu avancée, en négligeant l’apport de l’auto-protolyse de
l’eau, on obtient le tableau d’avancement :
HF + H2O−→F−+ H3O+KA=10−3,2 ≪1
EI c0 0
EF c h ≪c h ≪c
On obtient :
KA=h2
c⇔pH =pKA+pc
2=2,1
On doit vérifier les deux approximations :
1

— acide faiblement dissocié ; on doit être dans le do-
maine de prédominance de l’acide soit pH < pKA−
1=2,2 ; c’est bon ;
— apport de l’auto-protolyse de l’eau négligeable soit
pH < 6,5 ; c’est également bon.
R.2 Avec les mêmes hypothèses, on obtient pH =4,1 ce
qui est incorrect car on est dans le domaine de prédomi-
nance de la base. On doit revenir sur l’hypothèse de faible
dissociation ; le tableau d’avancement s’écrit alors :
HF + H2O−→F−+ H3O+KA=10−3,2 ≪1
EI c0 0
EF c−h h h
On obtient :
KA=h2
c−h⇔h=
−KA+qK2
A+4cKA
2pH =5,0
On est dans le domaine de prédominance de la base ; la
réaction est très avancée. On aurait pu faire cette hypo-
thèse et écrire h=csoit pH =pc =5; on retrouve la
formule valable pour l’acide fort. Cela illustre la loi de di-
lution d’Ostwald : un électrolyte est d’autant plus dissocié
que sa concentration est faible. Ici l’acide devient « fort ».
Dans le but de visualiser les différents régimes, la courbe
ci-dessous représente le pH en fonction de la concentration
de l’acide faible.
On constate, qu’à part pour de faibles concentrations
- auquel cas l’acide n’est pas vraiment utile - l’acide est
« faible » au sens faiblement dissocié.
Exercice 4 - Composition de l’eau de Javel
L’eau de Javel contient un mélange équimolaire de chlo-
rure de sodium (Na+,Cl−, concentration C0) et d’hypo-
chlorite de sodium (Na+,ClO−, concentration C0) en so-
lution aqueuse. On rappelle que HClO est un acide faible
de pKA=7,4.
Q.1 Quelle est l’espèce responsable des propriétés acido-
basiques de l’eau de javel ?
Q.2 Quelle est la composition, le pH de la solution pour
C0=1,0.10−2mol.l−1? Déterminer le degré d’avancement
α.
Corrigé
R.1 C’est bien évidemment la base faible ClO−.
R.2 En négligeant l’autoprotolyse de l’eau et en supposant
que la base est faiblement dissociée on a pour RPE non
quantitative :
ClO−+ H2O−→HClO + OH−KB=Ke
KA
EI c0 0 =10−6,6 ≪1
EF c−x≃c x x
On obtient :
pOH =pKB+pc
2=4,3 ⇔pH =pKe−pOH =9,7
On doit vérifier les deux approximations :
— base faiblement dissociée ; on doit être dans le do-
maine de prédominance de celle-ci soit pH > pKA+
1=8,4 ; c’est bon ;
— apport de l’auto-protolyse de l’eau négligeable soit
pH > 7,5 ; c’est également bon.
Enfin le taux de dissociation de la base vaut :
α=x
c=5.10−3≪1
c’est bien une « base faible ».
Exercice 5 - Acide oxalique
Q. Déterminer le pH d’une solution d’acide oxalique
H2C2O4noté dans la suite AH2, un diacide de pKAsuc-
cessifs pK1=1,2 et pK2=4,3 ; la concentration est
c=10−1mol.l−1.
Corrigé
R. On fait l’hypothèse que la première acidité est faible ;
on obtient pH = (pK1+pc)/2 =1,1 > pK1−1=0,2. L’hy-
pothèse n’est donc pas correcte. On ne peut pas négliger
la dissociation de la première acidité, mais on fait toujours
l’hypothèse que la seconde acidité n’intervient pas ; la RPE
non quantitative est ainsi :
AH2+ H2O−→AH−+ H3O+K1=10−1,2 ≪1
EI c0 0
EF c−h h h
2

On résout alors (à la main ou numériquement) :
K1=h2
c−h⇒pH =1,3
On a bien pH < pK2−1, la seconde acidité n’intervient
pas.
Pour visualiser le résultat quelquesoit la concentra-
tion initiale, les courbes suivantes présentent le pH et
les concentrations (avec domaines de prédominance) issus
d’une résolution numérique des équations en tenant compte
des deux acidités (mais en négligeant toujours l’autoproto-
lyse de l’eau) :
Exercice 6 - pH d’un mélange de deux acides
On considère une solution contenant HCl à une concen-
tration c0=1,0.10−1mol.l−1et de l’acide éthanoïque
CH3COOH à une concentration c1=c0(pour cet acide
on donne pKA=4,8).
Q.1 Calculer le pH de cette solution en précisant bien l’hy-
pothèse faite.
Q.2 Si c0baisse, le calcul précédent devient de moins en
moins valable. En supposant l’acide faiblement dissocié,
déterminer l’expression du pH en fonction de c1et tracer
la courbe correspondante.
Corrigé
R.1 On a un acide fort à forte concentration et un acide
faible qui, lorsqu’il est seul, est faiblement dissocié. On peut
raisonnablement penser que les ions H3O+apportés par
l’acide fort sont majoritaires par rapport à ceux apportés
par l’acide faible. On obtient ainsi immédiatement : pH=
pc0=1; l’hypothèse est correcte car pH< pKA−1=3,8 ;
l’acide éthanoïque n’est pratiquement pas dissocié. On a
[AH]≃c1et [A−] = KAc1/c0≈10−5mol.l−1.
R.2 Si c0baisse l’hypothèse précédente sera progressive-
ment de moins en moins correcte. On est obligé de tenir
compte de la RPE (non quantitative) :
AH + H2O−→A−+ H3O+KA≪1
EI c10c0
EF c1+c0−h h −c0h
On obtient alors :
KA=h(h−c0)
c1+c0−h
Cette équation du second degré en hse résout aisément et
on aboutit à :
h=c0−KA+q(KA−c0)2+4KA(c1+c0)
2
On peut alors tracer le pH et remarquer que l’on rejoint
les cas limites classiques (acide fort pour c0suffisamment
grand et acide faible sinon ; on peut visualiser également
le rapport [A−]/[H3O+]qui permet de valider l’hypothèse
faite à la première question s’il est suffisamment faible :
3

Enfin, en prenant la valeur de l’expression calculer pour
c0=0avec c1≫KA, on obtient la formule classique
h=√KAc1ou pH= (pKA+pc1)/2.
Exercice 7 - Dosage d’une solution de carbonate de
sodium
Une solution de carbonate de sodium (NaCO3) complète-
ment dissout comporte des ions Na+avec une concentra-
tion c0. On donne : pK1=pKA(H2CO3/HCO−
3) = 6,35 et
pK2=pKA(HCO−
3/CO2−
3) = 10,3. On dose v0=20 ml de
cette solution par de l’acide chlorhydrique de concentra-
tion c1=0,1 mol.l−1; on obtient ainsi le pH en fonction
du volume vd’acide versé.
Q.1 Décrire le matériel utilisé pour cette opération.
Quelles sont les réactions de dosage ? Calculer leurs
constantes. Déterminer c0.
Q.2 Tracer les domaines de prédominances des couples
acide-base. Quelles sont les espèces présentes pour v=0?
En déduire c0par le pH mesuré à v=0.
Q.3 Retrouver les différentes valeurs des constantes d’aci-
dité à partir de la courbe.
Q.4 Pour v=25 ml, justifier la valeur du pH.
Corrigé
R.1 C’est le cas classique du dosage d’une dibase faible
(CO2−
3) placée dans un bécher par un acide fort placé dans
une burette ; on suit le pH par une méthode potentiomé-
trique en plongeant une électrode de verre.
La première réaction de dosage est l’action de H3O+sur
la base la plus forte CO2−
3(on dose la première basicité) :
CO2−
3+ H3O+−→HCO−
3+ H2OK−1
2≫1
EI c0v0c1v ǫ
EF c0v0−c1v ǫ′c1v
La première équivalence (ve1 =10ml) correspond à la
(quasi-)disparition des ions carbonates soit pour : c0v0=
c1ve1 ⇔c0=c1ve1/v0=0,05 mol.l−1.
Après la première équivalence, la réaction de dosage est
l’action de H3O+sur la base la plus forte HCO−
3(on dose
la deuxième basicité ; on pose v′=v−ve1) :
HCO−
3+ H3O+−→H2CO3+ H2OK−1
1≫1
EI c0v0c1v′ǫ
EF c0v0−c1v′ǫ′c1v′
La deuxième équivalence définie par c0v0=c1(ve2 −ve1)
soit ve2 =20 ml correspond au dosage complet des ions
HCO−
3. Après la deuxième équivalence, on ajout un surplus
d’ions H3O+.
R.2 Pour pH<5,35, H2CO3domine, pour 7,35<pH<9,3,
HCO−
3et pour pH>11,3 ce sont les ions carbonate. Pour
v=0soit avant le dosage on a une solution de di-base
faible. En considérant que seule la première basicité inter-
vient et que la RPE (non quantitative) :
CO2−
3+ H2O−→HCO−
3+ OH−KB2
EI c00 0 =Ke
K2≪1
EF c−x≃c x x
est très peu avancée, on retrouve le résultat classique
pOH=(pKB2 +pc0)/2 soit pH=11,5. C’est ce qu’on peut
lire sur la courbe.
R.3 On utilise les demi-équivalence ; pour la première soit
v=ve1/2 on a CO2−
3] = [HCO−
3]soit pH=pK2. On lit en
effet sur la courbe pH≈10,4. De même pour la deuxième
équivalence soit v=ve1 +ve2/2, H2CO3] = [HCO−
3]soit
pH=pK1. On lit en effet sur la courbe pH≈6,3.
R.4 Après la deuxième équivalence, on est en présence
d’une solution d’acide chlorhydrique (fort) de concentra-
tion :
c′
1=c1v−2c0v0
v0+v
et donc de pH=pc′
1. Pour v=25ml, on trouve pH=1,95 ≈
2ce qu’on lit encore aisément sur la courbe.
Exercice 8 - Dosage d’une solution complexe
On réalise le titrage pHmétrique de v0=20 ml d’une
solution contenant c0=5.10−2mol.l−1d’acide chlorhy-
drique et une concentration c1d’ammoniac NH3, par une
4

solution d’hydroxyde de sodium de concentration c2=
4.10−2mol.l−1. On donne : pKA(NH+
4/NH3) = 9,2. On
obtient la courbe donnant le pH (de 0 à 14) en fonction
du volume pour v∈[0, 30]ml.
Q.1 Quelles sont les réactions du dosage ? En déduire c1.
Q.2 Où retrouve-t-on le pkAdu couple NH+
4/NH3?
Q.3 Justifier la valeur du pH pour v=30 ml.
Corrigé
R.1 Comme pour v=0,pH<pKA−1=8,2, on est très net-
tement dans le domaine de prédominance de NH+
4, ce qui
signifie c0> c1; la solution comporte initialement H3O+
en concentration c0−c1et NH+
4en concentration c1. On
étudie donc le dosage d’un mélange acide fort+acide faible
par une base forte.
La première réaction de dosage correspond à la RPQ de
l’acide le plus fort (H3O+) sur la base la plus forte (OH−) :
H3O++ OH−−→2H2OK−1
e=1014 ≫1
EI (c0−c1)v0c2v
EF (c0−c1)v0−c2v ǫ
La première équivalence (dosage du surplus d’acide fort)
correspond à (c0−c1)v0−c2ve1 =0; comme on lit ve1 =15
ml, on en déduit que c0−c1=3.10−2mol.l−1et donc que
c1=2.10−2mol.l−1.
Après la première équivalence, on dose la RPQ corres-
pond au dosage de l’acide le plus fort ie les ions ammonium
par la base la plus forte OH−(on note v′=v−ve1) :
NH+
4+ OH−−→H2O + NH3KA/Ke≫1
EI c1v0c2v′ǫ
EF c1v0−c2v′ǫ′c2v′
La deuxième équivalence correspond au dosage total des
ions ammonium soit : c2(ve2 −ve1) = c1v0; on retrouve la
valeur ve2 =25 ml que l’on peut lire sur la courbe.
Après la deuxième équivalence on ajoute seulement un
surplus d’ions OH−.
R.2 Le pKAdu couple peut se lire sur la deuxième demi-
équivalence pour c2v′=c1v0/2 soit pour v=20 ml lorsque
[NH+
4] = [NH3]; on a alors pH=pKA=9, 3 que l’on lit
sur la courbe.
R.3 Pour v=30 ml, on a simplement une solution de base
forte de concentration :
c′
2=c2(v−ve2)
v+v0
En utilisant alors pOH =pc′
2on obtient bien pH=11,6
que l’on peut lire sur la courbe.
5
1
/
5
100%