Lire l'article complet

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 1 - janvier-février 2009
23
23
dossier thématique
* Centre de recherche
des Cordeliers, Inserm U872,
équipe 8, université Paris-VI,
pathologies nutritionnelles
et métaboliques : diabète,
obésité, Paris.
Développement du pancréas : les points clés
Depuis une vingtaine d’années, de nombreuses études
ont été consacrées au développement du pancréas. On
comprend mieux aujourd’hui comment il se développe
et quels sont les mécanismes qui régulent ce proces-
sus. Le pancréas dérive de l’endoderme et se forme
à partir de bourgeons pancréatiques émergeant du
tube digestif fœtal. La spéci cation de chaque type
cellulaire est contrôlée par la mise en place de pro-
grammes génétiques dépendant de l’expression de
facteurs de transcription. Parmi ceux-ci, Pdx-1 et Ptf1a
sont deux facteurs clés exprimés dans les cellules pro-
génitrices. Quand celles-ci s’engagent dans la voie de
di érenciation exocrine, elles perdent l’expression de
Pdx-1. À l’inverse, la voie de di érenciation endocrine
Nutrition fœtale et développement
des cellules bêta : rôle des glucocorticoïdes
Fetal nutrition and beta cells development: the role of glucocorticoids
Bertrand Blondeau, Bernadette Breant*
Le diabète de type 2 peut être programmé pendant la vie intra-
»
utérine, en particulier par l’état nutritionnel.
La période sensible de développement des cellules bêta est la vie
»
fœtale.
Chez les rongeurs, l’exposition à des taux excessifs de glucocorticoïdes
»
inhibe le développement des cellules bêta, par l’intermédiaire du
récepteur GR dans les cellules précurseurs pancréatiques.
Le récepteur GR est présent dans le pancréas fœtal humain en
»
développement, dans les cellules qui expriment la machinerie
protéique de diff érenciation en cellules bêta.
La surexposition à des taux excessifs de glucocorticoïdes au
»
cours de la vie fœtale chez l’homme pourrait-elle endommager le
développement des cellules bêta ?
Mots-clés : Pancréas – Développement – Nutrition – Glucocorticoïdes
– Cellules précurseurs – Souris mutantes.
Keywords: Pancreas – Development – Nutrition – Glucocorticoids –
Precursor cells – Mutant mice.
Points forts
Nutrition et croissance fœtale
La croissance fœtale est un processus complexe, dyna-
mique et qui dépend étroitement de l’apport continu
de nutriments par l’organisme maternel. Les grandes
variations de croissance fœtale que l’on peut observer
chez l’homme résultent de l’in uence des facteurs envi-
ronnementaux sur le programme génétique de déve-
loppement. En e et, les organes se développent durant
des fenêtres de croissance di érentes, et on comprend
aisément combien une croissance inadéquate pendant
ces périodes critiques peut conduire à des défauts irré-
versibles. Des études menées depuis presque deux
décennies chez l’homme ont montré des associations
fortes entre croissance fœtale et apparition de maladies
dégénératives chez l’individu devenu adulte. De fait, les
nouveau-nés de faible poids à la naissance présentent, à
l’âge adulte, un risque élevé de développer des maladies
cardio-vasculaires et, surtout, un diabète de type 2 avec
diminution de la sensibilité à l’insuline (1, 2). De ces obser-
vations a émergé le concept de la programmation fœtale
des maladies de l’adulte. Les données cliniques permet-
tent de penser que la malnutrition ou la sous-nutrition
fœtale conduisant à un retard de croissance intra-utérin
pourrait a ecter la programmation de la régulation post-
natale du métabolisme du glucose. Le risque accru de
développer une telle maladie peut s’expliquer soit par
un défaut de développement des cellules bêtapancréa-
tiques, soit par la constitution d’une insulinorésistance,
soit par l’association de ces deux composantes.
De nombreux modèles animaux de retard de crois-
sance intra-utérin ont été développés pour tenter
d’apporter des éléments décisifs de compréhension.
Ces modèles mettent en œuvre soit des restrictions
vasculaires (ligature des artères utérines), soit des res-
trictions alimentaires chez la femelle gestante (restric-
tion protéique ou calorique globale). Dans cette revue,
nous nous restreindrons aux modèles hypocaloriques
développés chez les rongeurs (le rat et la souris), et
évoquerons les hypothèses auxquelles ils ont conduit
et les mécanismes impliqués, ainsi que les applications
potentielles chez l’homme.
>>>

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 1 - janvier-février 2009
24
24
dossier thématique
implique l’acquisition de facteurs tels que Ngn3, et
la spéci cation en cellule bêta exprimant l’insuline
requiert l’expression de Pdx-1 et d’autres facteurs tels
que Nkx2.2 (3). Dans ce contexte, il paraît évident que
des événements qui contrôleraient l’expression de ces
facteurs contrôleraient également le devenir des cellules
en cours de di érenciation.
La vie fœtale, une période cible
et le pancréas, un tissu cible
A n de tester l’hypothèse d’un défaut primitif de déve-
loppement des cellules bêta fœtales, nous avons conçu
chez le rat un modèle de sous-nutrition impliquant une
réduction globale de l’apport alimentaire maternel de
la dernière semaine de grossesse jusqu’au sevrage. La
masse de cellules bêta peut être facilement quanti-
ée par morphométrie quantitative, une méthode qui
associe la détection des cellules bêta sur des coupes de
pancréas par leur expression de l’insuline et leur quan-
ti cation grâce à un logiciel. Dans ce modèle de restric-
tion alimentaire, les fœtus ont un retard de croissance
irréversible et présentent une diminution de la masse
de cellules bêtapancréatiques, qui persiste à l’âge adulte
et conduit nalement à une intolérance au glucose
(4, 5). De plus, les femelles adultes issues de ce modèle
sont incapables d’adapter leur masse pancréatique à
la gestation (6), et leurs fœtus présentent eux aussi un
défaut de développement des cellules bêta (7).
Des modèles équivalents ont été développés plus
récemment chez la souris. Un régime hypocalorique
au cours de la dernière semaine de gestation entraîne
chez la souris un faible poids à la naissance, qui se nor-
malise ensuite. Cependant, une intolérance au glucose
apparaît chez le rongeur adulte caractérisée par un
défaut majeur de sécrétion d’insuline malgré une masse
de cellules bêta normale (8).
Ces observations sont en faveur d’un rôle de la nutrition
intra-utérine dans la programmation du développement
des cellules bêta, et de l’altération de l’homéostasie
glucidique chez les rongeurs.
Surexposition aux glucocorticoïdes
in utero, développement des cellules bêta :
une voie de programmation du diabète ?
Parmi les diverses réactions physiologiques induites par
la malnutrition, des données récentes ont mis en avant
le rôle des glucocorticoïdes (GC) libérés sous l’e et du
stress. Chez les rongeurs, l’exposition du fœtus à des
concentrations excessives de GC maternels conduit à
un faible poids à la naissance et à l’altération ultérieure
de la tolérance au glucose et à l’hypertension (9, 10).
Ces hormones pourraient-elles intervenir en modi ant
l’équilibre des facteurs de transcription gouvernant le
développement des cellules bêta et ainsi conduire, si
leur nombre est insu sant, à une altération de l’équi-
libre glycémique chez l’adulte ?
C’est ce que nous avons exploré dans le modèle de res-
triction calorique. La restriction alimentaire maternelle
induit une augmentation des concentrations maternel-
les et fœtales de corticostérone, qui, à son tour, réduit
la masse des cellules bêta et leur di érenciation chez
le fœtus ( gure 1, à gauche). Plus généralement, chez
le rat nourri normalement, la diminution de la masse
des cellules bêta est observée dans des conditions de
surexposition aux GC, alors que le nombre de ces cel-
lules augmente dans des conditions d’hypocorticos-
téronémie (11) [ gure 1, à droite]. La diminution de la
masse des cellules bêta est associée à une expression
moindre du facteur de transcription Pdx-1, spéci que
des cellules endocrines pancréatiques précurseurs mais
aussi marqueur des cellules bêta di érenciées.
En accord avec ces données chez l’animal entier, nous
avons montré, par des études in vitro, que les GC
favorisent la di érenciation des cellules précurseurs
du pancréas en cellules exocrines, mais répriment leur
di érenciation en cellules endocrines. Ces variations
suivent les variations observées dans le niveau d’ex-
pression des facteurs de transcription qui régissent la
di érenciation pancréatique, et en particulier la dimi-
nution de l’expression du facteur de transcription Pdx-1
(12) [ gure 2]. L’activation du récepteur aux GC par le
ligand est donc impliquée dans la genèse des altérations
induites par la nutrition hypocalorique in utero et o re
une voie de régulation du développement des cellules
bêta jusqu’alors inconnue.
Ces données suggèrent que les GC pourraient pro-
grammer la masse de cellules bêta chez les rongeurs,
et ainsi perturber plus tard l’homéostasie glucidique
en modi ant l’équilibre des facteurs de transcription
qui régulent la di érenciation du pancréas.
Programmation ou induction du diabète
par les glucocorticoïdes ?
Un dysfonctionnement de la réponse au stress est
soupçonné dans l’étiologie de plusieurs désordres
métabo liques, comme le diabète de type 2. Ce dysfonc-
tionnement peut résulter de l’exposition fœtale à un
environnement perturbé ou bien être acquis lors de la vie

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 1 - janvier-février 2009
25
25
Nutrition fœtale et développement des cellules bêta : le rôle des glucocorticoïdes
adulte. Dans ce contexte, le rôle du récepteur des GC (GR),
un facteur de transcription activé par les hormones GC,
se pose de manière aiguë. Chez l’homme, il est clair que
l’exposition prénatale à des taux élevés de ces hormones,
par exemple lors de traitements à la prednisone ou dans
le cas d’un syndrome de Cushing chez la mère, est asso-
ciée à un retard de croissance intra-utérin (13, 14), mais
les conséquences métaboliques plus tardives ne sont
pas connues. Nos travaux passés montrent que, e ec-
tivement, un stress élevé qui s’exerce sur les embryons
des rongeurs a des conséquences à long terme et que
ces animaux, une fois adultes, présentent une intolérance
au glucose. Nous venons de voir que ce phénomène est
dû à l’action des GC, libérés en réponse au stress, sur
le pancréas. Puisque nous savons que ces hormones
agissent à de multiples niveaux, seule une dissection
génétique de la fonction du gène du GR, par mutagenèse
conditionnelle, couplée à des analyses anatomiques et
métaboliques permet de comprendre comment ces
hormones favorisent l’apparition du diabète. Dans cette
optique, nous analysons et développons chez la souris
des modèles dans lesquels la signalisation par les GC est
abolie par l’invalidation du gène GR dans une population
de cellules données, les cellules bêta matures, les cellules
précurseurs pancréatiques ou l’organisme entier.
Les mutants GR null/null
L’analyse du phénotype pancréatique de souris mutan-
tes GR null/null dépourvues de GR dans l’ensemble de
l’organisme a montré que le GR est impliqué dans la
survie et l’organisation du tissu pancréatique. Le GR
n’est pas nécessaire aux phases précoces du dévelop-
pement, mais son dosage est critique pour moduler
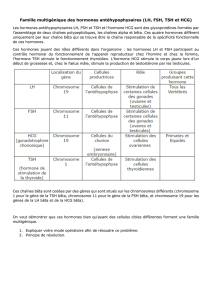
Figure 1. Le développement des cellules bêta fœtales est sensible aux glucocorticoïdes.
L’exposition à une dose excessive de glucocorticoïdes (nutrition hypocalorique ou nutrition normale avec traitement maternel à la dexaméthasone [Dex]) diminue le développement des cellules bêta. À
l’inverse, des taux faibles de glucocorticoïdes (surrénalectomie maternelle + traitement à la métopyrone), favorisent le développement des cellules bêta.
Surrénalectomie
+ métopyrone
+Dex
Rat nourri normalement
Glucocorticoïdes
Glucocorticoïdes
Nutrition normale
+ intolérance au glucose
nouveau-né
adulte
Régime hypocalorique
(sous-nutrition fœtale)
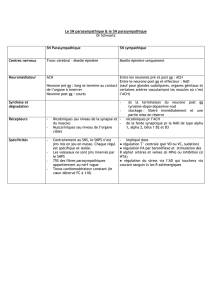
Figure 2. Schéma simpli é des principaux facteurs de transcription impliqués dans le développement pancréatique.
Les mutants dépourvus de récepteur aux glucocorticoïdes dans tout le pancréas ou dans les cellules bêta matures
identi ent une cellule précurseur à vocation endocrine pré-bêta comme cellule cible des glucocorticoïdes ( èche).
PP : polypeptide pancréatique.
Ptf1a/Pdx-1
Bourgeons
pancréatiques
Pdx-1 Insuline
Glucocorticoïdes
Ptf1a Ngn3
alpha bêta delta PP
Cellule
exocrine

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 1 - janvier-février 2009
26
26
dossier thématique
l’expansion de la masse des cellules bêta aux stades
fœtaux plus tardifs. En e et, de manière intéressante,
les fœtus hétérozygotes GR +/null présentent un dou-
blement de la masse des cellules bêta, qui persiste à
l’âge adulte (15).
Les mutants d’inactivation spéci que du GR
dans le pancréas ou les cellules bêta
Les souris chez lesquelles le GR a été invalidé dans les
cellules pancréatiques précurseurs (souris GR
PdxCre
ou
GRpanc–) ont une masse de cellules bêta doublée et un
nombre d’îlots très nettement augmenté par rapport
aux souris témoins GR+ (ou GRlox/lox), alors que la masse
de cellules alpha (produisant le glucagon) n’est pas
a ectée ( gure 3). À l’inverse, les souris dont le GR est
invalidé dans les cellules bêta matures (souris GRRIPCre, ou
GRbêta–) ne présentent aucune altération de la masse des
cellules bêta et alpha. Ces résultats identi ent comme
cellule cible des GC la cellule précurseur pancréatique
à vocation endocrine ( gure 2, èche), et montrent
que, en l’absence de signalisation GC dans les cellules
précurseurs, celles-ci se di érencient préférentiellement
en cellules bêta (12).
Ces modèles de souris constituent aussi des outils pré-
cieux pour savoir si tous les e ets de la sous-nutrition
décrits sur le développement des cellules bêta font
intervenir l’activation du gène du récepteur aux GC.
En analysant la masse de cellules bêta chez les fœtus
normalement nourris ou sous-alimentés à 50 % issus de
femelles GR+ porteuses pour moitié de fœtus GRpanc– et
pour moitié de fœtus GR+, nous avons démontré que
les e ets délétères de la sous-nutrition in utero sur le
développement des cellules bêta nécessitaient la pré-
sence d’un GR fonctionnel dans les cellules précurseurs
pancréatiques.
Glucocorticoïdes et développement
des cellules bêta chez l’homme ?
Chez l’homme, les études des conséquences de l’expo-
sition prénatale aux GC sont rares. Il est bien connu que
l’exposition prénatale à la prednisone et le syndrome de
Cushing maternel sont associés à une fréquence plus
élevée de retard de croissance intra-utérin (13, 14). Le
poids à la naissance est donc sensible aux GC maternels,
à la fois chez les rongeurs et chez l’homme. Nos études
chez les rats et les souris montrent que l’expansion de la
masse de cellules bêta est sensible aux GC pendant une
fenêtre critique de son développement. Nos résultats
sur des spécimens de pancréas de fœtus humain à des
stades précoces de grossesse indiquent que le GR est
exprimé très tôt, dès 6 semaines de développement
dans le mésenchyme et à partir de 10 semaines dans
les cellules épithéliales précurseurs exprimant le fac-
teur de transcription Pdx-1, au moment où ces cellules
commencent à exprimer l’insuline (16). La réponse aux
hormones GC est donc présente dans le pancréas fœtal
humain dans les cellules qui expriment les éléments
de la machinerie de di érenciation pancréatique en
cellules bêta. Il est intéressant de noter qu’au fur et à
mesure que le développement pancréatique progresse,
les cellules bêta sont les seules cellules pancréatiques
à exprimer le GR et sont donc seules susceptibles de
répondre à ces hormones, en termes de développement
ou de sécrétion d’insuline. Dans quelle mesure l’expo-
sition du fœtus humain à un excès de GC pourrait-elle
moduler la croissance des cellules bêta au cours d’une
fenêtre sensible de son développement et être associée
à l’intolérance au glucose et à un dé cit d’insulinosécré-
tion chez ces individus devenus adultes ? La question
est à l’étude.
■
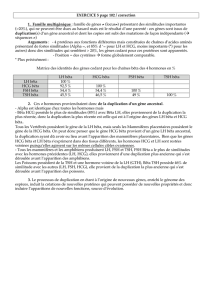
Figure 3. L’invalidation du récepteur aux glucocorticoïdes (GR) précocement dans le développement pancréatique (souris GR-PdxCre) augmente la masse de cellules bêta chez l’adulte, sans a ecter la
masse des cellules alpha.
À gauche, marquage immunohistochimique pour l’insuline (en marron) sur des pancréas témoins (GR-lox/lox) ou mutants.
** di érence signi cative, p < 0,01, par rapport aux témoins.
GRlox GRPdxCre
GRPdxCre GRPdxCre
GRlox
Masse de cellules
bêta (mg)
30,4
**
2
0,2
1
0
Masse de cellules
alpha (mg)
GRlox

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 1 - janvier-février 2009
27
27
Nutrition fœtale et développement des cellules bêta : le rôle des glucocorticoïdes
Nouvelles de l’industrie pharmaceutique
Communiqués des conférences de presse, symposiums, manifestations, organisés par l’industrie pharmaceutique
1. Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM.
Type 2 (non-insulin-dependent) diabetes mellitus, hypertension
and hyperlipidaemia (syndrome X): relation to reduced foetal
growth. Diabetologia 1993;36:62-7.
2. Hales CN, Barker DJ, Clark PM et al. Foetal and infant
growth and impaired glucose tolerance at age 64. BMJ
191;303:1019-22.
3. Murtaugh LC. Pancreas and beta-cell development: from the
actual to the possible. Development 2007;134:427-38.
4. Garofano A, Czernichow P, Breant B. In utero undernutrition
impairs rat beta-cell development. Diabetologia 1997;40:1231-4.
5. Garofano A, Czernichow P, Breant B. Eff ect of ageing on
beta-cell mass and function in rats malnourished during the
perinatal period. Diabetologia 1999;42:711-8.
6. Blondeau B, Garofano A, Czernichow P, Breant B. Age-
dependent inability of the endocrine pancreas to adapt to
pregnancy: a long-term consequence of perinatal malnutrition
in the rat. Endocrinology 1999;140:4208-13.
7.
Blondeau B, Avril I, Duchene B, Breant B. Endocrine pancreas
development is altered in foetuses from rats previously showing
intra-uterine growth retardation in response to malnutrition.
Diabetologia 2002;45(3):394-401.
8.
Jimenez-Chillaron JC, Hernandez-Valencia M, Reamer C et
al. Beta-cell secretory dysfunction in the pathogenesis of low
birth weight-associated diabetes: a murine model. Diabetes
2005;54(3):702-11.
9. Seckl JR. Glucocorticoids, feto-placental 11 beta-hydroxys-
teroid dehydrogenase type 2, and the early life origins of adult
disease. Steroids 1997;62:89-94.
10. Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR.
Glucocorticoid exposure in late gestation permanently pro-
grams rat hepatic phosphoenolpyruvate carboxykinase and
glucocorticoid receptor expression and causes glucose intole-
rance in adult off spring. J Clin Invest 1998;101:2174-81.
11. Blondeau B, Lesage J, Czernichow P, Dupouy JP, Breant B.
Glucocorticoids impair foetal beta-cell development in rats.
Am J Physiol Endocrinol Metab 2001;281:E592-9.
12. Gesina E, Tronche F, Herrera P et al. Dissecting the role
of glucocorticoids on pancreas development. Diabetes
2004;53:2322-9.
13.
Reinisch JM, Simon NG, Karow WG, Gandelman R. Prenatal
exposure to prednisone in humans and animals retards intrau-
terine growth. Science 1978;202:436-8.
14. Dalziel SR, Walker NK, Parag V et al. Cardiovascular
risk factors after antenatal exposure to betamethasone:
30-year follow-up of a randomised controlled trial. Lancet
2005;365:1856-62.
15.
Gesina E, Blondeau B, Milet A et al. Glucocorticoid signal-
ling aff ects pancreatic development through both direct and
indirect eff ects. Diabetologia 2006;49(12):2939-47.
16.
Phan-Hug F, Guimiot F, Lelièvre V et al. Potential role of
glucocorticoid signaling in the formation of pancreatic islets in
the human fetus. Pediatr Res 2008. [Epub ahead of print].
Références
Changer l’avenir des enfants
atteints de diabète
Dans le monde, environ 246 millions de per-
sonnes sont atteintes de diabète. Ce chi re
devrait atteindre 380 millions en 2025 ; plus
de 80 % des cas devraient alors concerner les
pays à revenu faible ou moyen. Cette a ec-
tion est la quatrième cause de mortalité par
maladie. Actuellement, 440 000 enfants de
moins de 14 ans vivent avec un diabète de
type 1, dont 250 000 dans les pays en voie de
développement et 70 000 dans des condi-
tions désespérées.
Novo Nordisk a lancé, à l’occasion du 60e anni-
versaire de la Déclaration universelle des
droits de l’homme des Nations unies, un
programme sur 5 ans d’accès gratuit à l’in-
suline et au traitement du diabète pour les
enfants des pays les plus pauvres de la pla-
nète. Le programme “Changer l’avenir des
enfants atteints de diabète” débute en 2009
en Ouganda, en Tanzanie, en Guinée Conakry
et en République démocratique du Congo.
Environ 38 000 enfants africains de moins de
14 ans sont touchés par le diabète de type 1.
Dans les pays pauvres, comme ceux de l’Afri-
que subsaharienne, ces enfants sont parti-
culièrement vulnérables, et leur espérance
de vie est inférieure à un an, alors que dans
les pays développés ils pourraient mener une
vie presque normale. Le manque d’insuline
est la cause de mortalité la plus fréquente
chez les enfants diabétiques. La situation
est très inquiétante dans les pays à revenu
faible ou moyen, où de nombreux petits dia-
bétiques meurent en raison d’un diagnostic
trop tardif, d’un accès insu sant aux soins et
à l’éducation au diabète ; la famille n’a pas
les moyens de payer un traitement médical
approprié. Les données disponibles montrent
que de nombreux enfants meurent peu après
le diagnostic ou contrôlent mal leur diabète
et développent des complications précoces
sévères (cécité, insu sance rénale, lésions
neurologiques).
Ce programme fonctionnera selon un concept
en étoile (création de centres satellites autour
des structures de soins existantes) et aura
pour objectif de mettre en place des solutions
à long terme pour la distribution de l’insuline
et la prise en charge durable de la maladie
auprès de l’ensemble des diabétiques des
pays les plus pauvres de la planète. L’idée
est de collaborer avec le plus grand nombre
de partenaires locaux (gouvernements et
associations de diabétiques), les antennes
régionales de la Fédération internationale
du diabète et les grands leaders d’opinion.
L’amélioration des infrastructures sanitaires
des pays participants contribuera à faire vivre
le programme au-delà des 5 années de mise
en place.
MP
>>>
1
/
5
100%